



Child with a history of multiple fractures
The patient, an 8-year-old male who recently immigrated to the United States from El Salvador, initially presented to the emergency department (ED) for a cough. The next day, he went to the general pediatrics clinic for follow-up and was noted to have a significant history of recurrent fractures.


Figure 1



Table
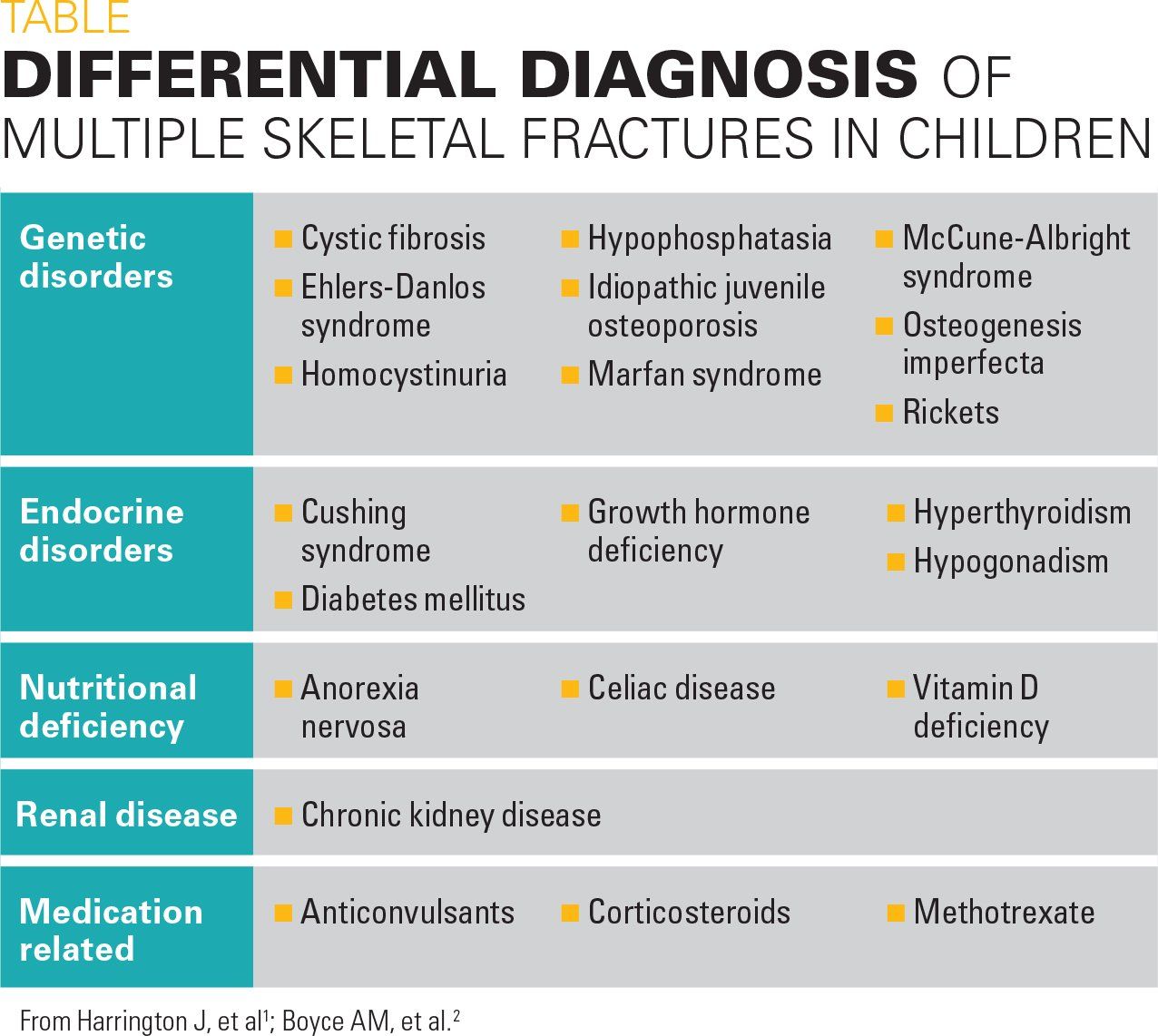


Figure 2
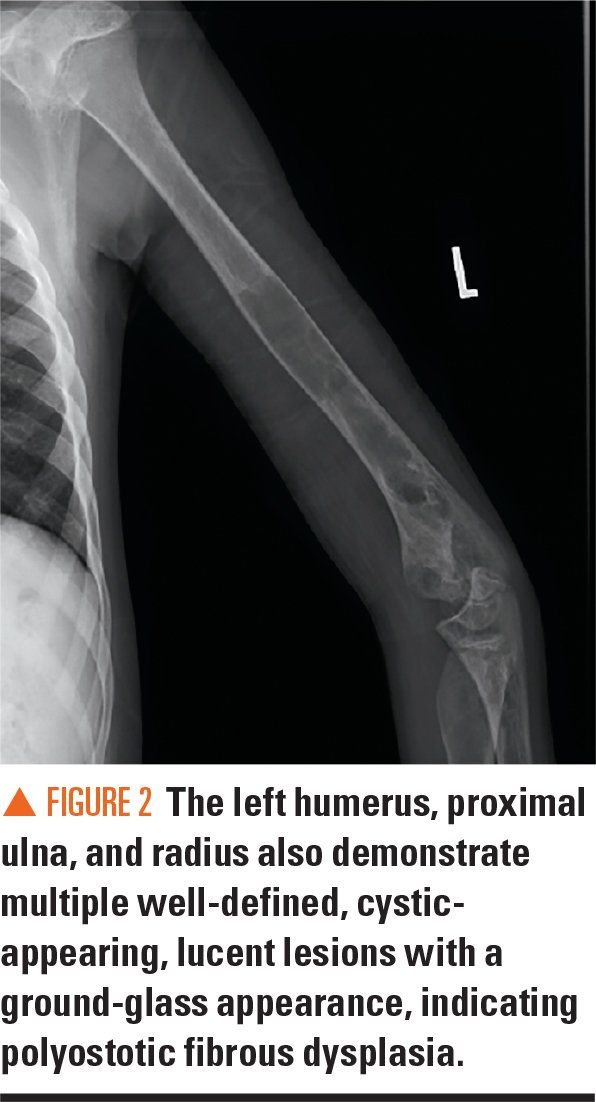


Figure 3
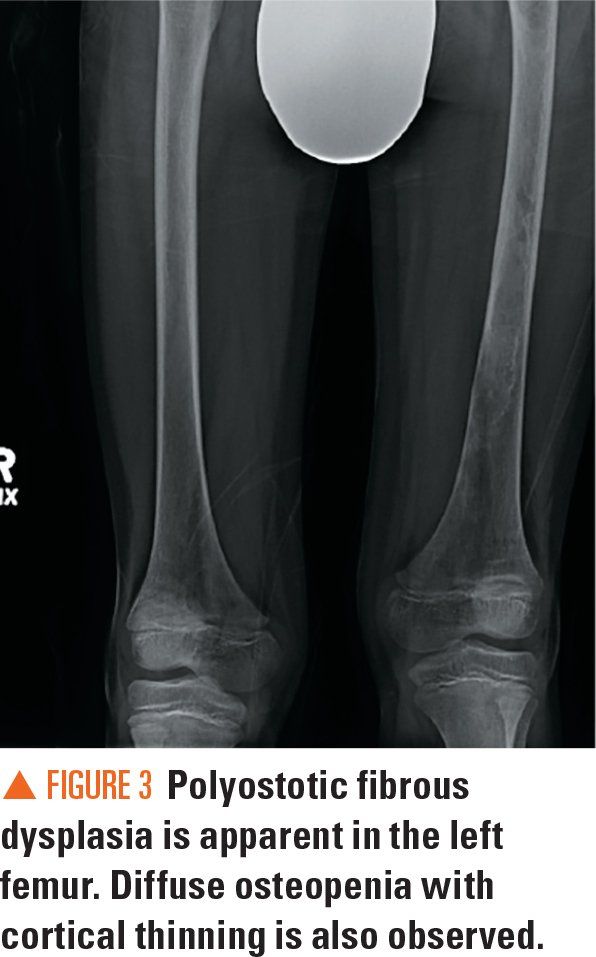


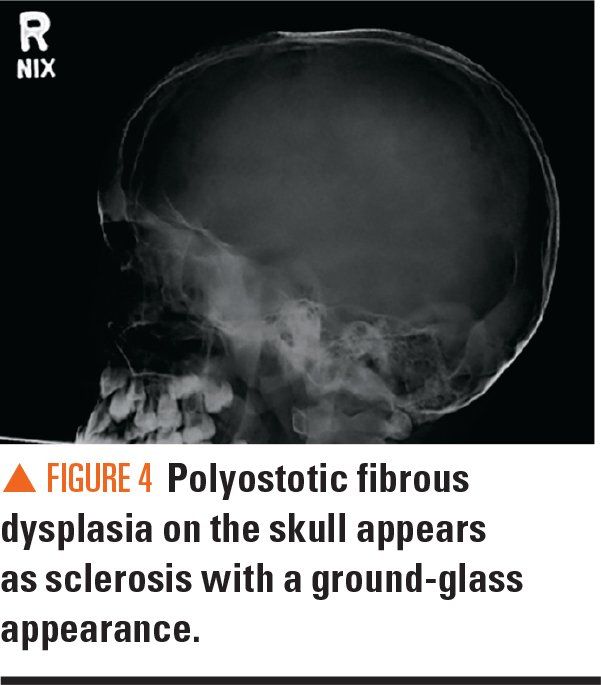
Figure 4
The case
The patient, an 8-year-old male who recently immigrated to the United States from El Salvador, initially presented to the emergency department (ED) for a cough. The next day, he went to the general pediatrics clinic for follow-up and was noted to have a significant history of recurrent fractures.
The father reported his son had had 7 fractures of the upper extremities beginning at age 3 years (on later detailed inquiry, 10 fractures were reported: 2 fractures of the left arm and 8 fractures of the right arm), with the latest fracture occurring approximately 3 months ago to the right humerus. The fractures were sustained with minimal force. The family reported that their child was diagnosed with osteogenesis imperfecta in their home country. The ED had discharged the patient with the same diagnosis.
Physical examination
On physical exam, the patient was alert, cooperative, and in no apparent distress, with unremarkable vital signs. His height was 127.7 cm (40.5th percentile for age and sex) and weight was 23 kg (18.2th percentile for age and sex). There was a sizeable callus and bowing deformity of the right forearm, as well as leg length discrepancy (right longer than left). The patient also was noted to have hyperpigmented macular skin lesions.
Consultations and additional studies
The patient was referred by the general pediatrics clinic to Pediatric Endocrinology. Further history revealed that he also had primary dentition issues, including losing his primary teeth early at the age of 4 years. Findings on physical exam included that the sclerae were white and not blue; the hyperpigmented macules were located on the left posterior thorax and left side of the neck and spared the midline; and scant pubic hair was present.
Initial laboratory test results had included normal values for serum calcium (9.6 mg/dL), magnesium (2.1 mg/dL), phosphorus (4.5 mg/dL), and 25-hydroxy vitamin D (32.3 ng/mL). A serum immunoglobulin (Ig) E was within normal range. Parathyroid hormone intact (PTHi) was slightly elevated to 61 pg/mL (normal range, 9-59 pg/mL), and osteocalcin was elevated to 217 ng/mL (normal range, 47-142 ng/mL). Based on the clinical findings, further laboratory tests were ordered: insulin-like growth factor 1 (IGF-1) was 118 ng/mL (normal, 52-391 ng/mL); insulin-like growth factor binding protein 3 (IGF-BP3) was 3.6 mg/L (normal, 1.2-6.4 mg/L); total testosterone was 17 ng/dL (normal, <13 ng/dL); thyroid stimulating hormone (TSH) was <0.01 μIU/mL (normal, 0.35-4.94 μIU/mL); total thyroxine (T4) was 12.87 μg/dL (normal, 4.87-11.72 μg/dL); and free T4 was 2.27 ng/dL (normal, 0.7-1.48 ng/dL).
Bilateral radiographs of the forearm and humerus revealed numerous lucent lesions and areas of sclerosis with a bowing defect of the right humerus. A skeletal survey revealed a healing pathologic midshaft fracture in the right humerus. In addition, numerous right and left humeral, ulnar, and radial well-circumscribed, centrally lucent lesions with some areas of sclerosis were visualized. There was large distortion and mottled sclerosis of the left iliac wing. These radiograph findings were consistent with polyostotic fibrous dysplasia.
Discussion
The differential diagnosis for conditions leading to increased risk of fragility fractures in children is extensive, thus a thorough history is essential in narrowing the potential etiologies (Table).1,2 Clinicians should always consider nonaccidental trauma as a potential cause for multiple healing fractures, especially when there is a discrepancy between the history provided by the caregivers and physical exam findings. In the present case, the history did not raise suspicion for nonaccidental trauma, and the multiple historical and physical findings strongly suggested a genetic etiology.
The patient was initially presumed to have osteogenesis imperfecta (OI), which is the most common cause of primary osteoporosis, leading to bone fragility and fractures due to defects in the quality or quantity of collagen type I.3 Multiple different subtypes of OI have been identified, with a range of severity and clinical presentations. The OI type I is the most common and mildest form of the disease, and is characterized by blue-tinted sclerae, normal to slightly below-normal stature, and no dental involvement. The OI type II patients usually do not survive beyond the newborn period, whereas OI type III is characterized by severe disease with bony deformities apparent at birth. Both OI type IV and type V are moderate in severity, but type V can be distinguished by hyperplastic calluses at fracture sites.
McCune-Albright syndrome (MAS) is also a rare disorder that can be easily mistaken for OI based on a history of multiple fractures. The prevalence is between 1/100,000 and 1/1,000,000. The classic triad of MAS consists of fibrous dysplasia, precocious puberty, and café au lait spots, However, multiple hyperfunctioning endocrinopathies are also seen in MAS, including hyperthyroidism, hypercortisolism, excess growth hormone production, renal phosphate wasting with possible rickets, and Cushing syndrome.4,5 Because the patient had polyostotic fibrous dysplasia, café au lait spots, and hyperthyroidism, a diagnosis of MAS was made.
An activation of the Gsα protein encoded by the GNAS gene is the underlying genetic defect that leads to MAS.5 Melanocyte stimulating hormone (MSH), luteinizing hormone (LH), TSH, growth-hormone-releasing hormone (GHRH), and adrenocorticotropic hormone (ACTH) all use G-protein pathways for signaling. Thus, a constitutional activation of the G-protein pathways in MAS leads to increased cyclic adenosine monophosphate (cAMP) and increased production of melanin, estradiol, testosterone, thyroid hormone, growth hormone, and cortisol.
Because MAS is not vertically transmitted, patients are considered to be somatic mosaics; thus, the diagnosis is based on clinical findings, with no definitive genetic testing available.
Patient outcome
The patient was started on methimazole for hyperthyroidism. Bisphosphonate therapy was considered but not pursued initially, given studies are inconclusive regarding its effect on the progression of fibrous dysplasia, and given he was not experiencing significant bone pain when first seen in the clinic.
The patient has continued to be closely monitored for development of other potential endocrinopathies, which he has not developed, and he appears to be progressing through puberty6 in a normal timeline.
Lessons for the clinician
The differential diagnosis for a patient with a history of multiple fractures is expansive, and clinicians may see a patient with MAS and easily mistake it for a diagnosis of osteogenesis imperfecta. Diligent history taking, a thorough physical exam, focused laboratory studies, and careful review of imaging were key to unraveling this diagnosis.
In order to minimize the progression of fibrous dysplasia in MAS patients, correction of thyroid dysfunction, as well as proper supplementation of calcium and vitamin D, may be required. The potential benefits of bisphosphonate treatment have been shown to yield positive results in some MAS patients.7,8 However, no universal recommendation for bisphosphonate use is currently available.
References:
1. Harrington J, Sochett E. The child with multiple fractures, what next? Pediatr Clin North Am, 2015;62(4):841-855.
2. Boyce AM, Gafni RI. Approach to the child with fractures. J Clin Endocrinol Metab. 2011;96(7):1943-1952.
3. Van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature, and severity assessment. Am J Med Genet A. 2014;164A(6):1470-1481. Erratum in: Am J Med Genet A. 2015:167A(5):1178.
4. Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J Rare Dis. 2012;7(suppl 1):S4.
5. Salpea P, Stratakis CA. Carney complex and McCune Albright syndrome: an overview of clinical manifestations and human molecular genetics. Mol Cell Endocrinol. 2014;386(1-2):85-91.
6. Fuqua J.S. Treatment and outcomes of precocious puberty: an update. J Clin Endocrinol Metab. 2013;98(6):2198-2207.
7. Majoor BC, Appelman-Dijkstra NM, Fiocco M, van de Sande M.A, Dijkstra PS, Hamdy NA. Outcome of long-term bisphosphonate therapy in McCune-Albright syndrome and polyostotic fibrous dysplasia. J Bone Miner Res. 2017;32(2):264-276.
8. Wang Y, Wang O, Jiang Y, et al. Efficacy and safety of bisphosphonate therapy in McCune-Albright syndrome and related polyostotic fibrous dysplasia: a single-center experience. Endocr Pract. November 1, 2018. Epub ahead of print.















Boy presents with fatigue, minimal responsivity, and diffuse muscle weakness
August 7th 2024An 11-year-old boy with a history of asthma and allergic rhinitis presented to the emergency department (ED) with worsening fatigue, minimal responsivity to external stimuli, and diffuse muscle weakness for 2 months.

A 9-year-old boy presents with neck mass
July 3rd 2024A 9-year-old boy was seen for follow-up of a neck mass noted several years earlier. He first presented with this finding at 3 years of age, when during an otherwise unremarkable examination, he became upset, and a protuberant swelling was noted in the anterior aspect of the right side of his neck. What's the diagnosis?

