


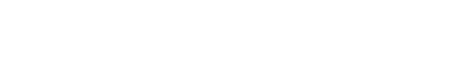
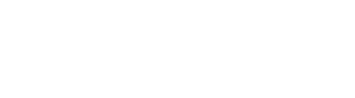

Infant’s leg swelling could be malignancy
A 5-month-old previously healthy, full-term female presented to a pediatric emergency department with 2 weeks of left leg swelling. Her parents denied any history of trauma, pain, fevers, weight loss, and easy bruising or bleeding, and family history was negative for cancer. The patient had been feeding and eliminating well.


Figures 1A,1B, 2A, and 2B


Table


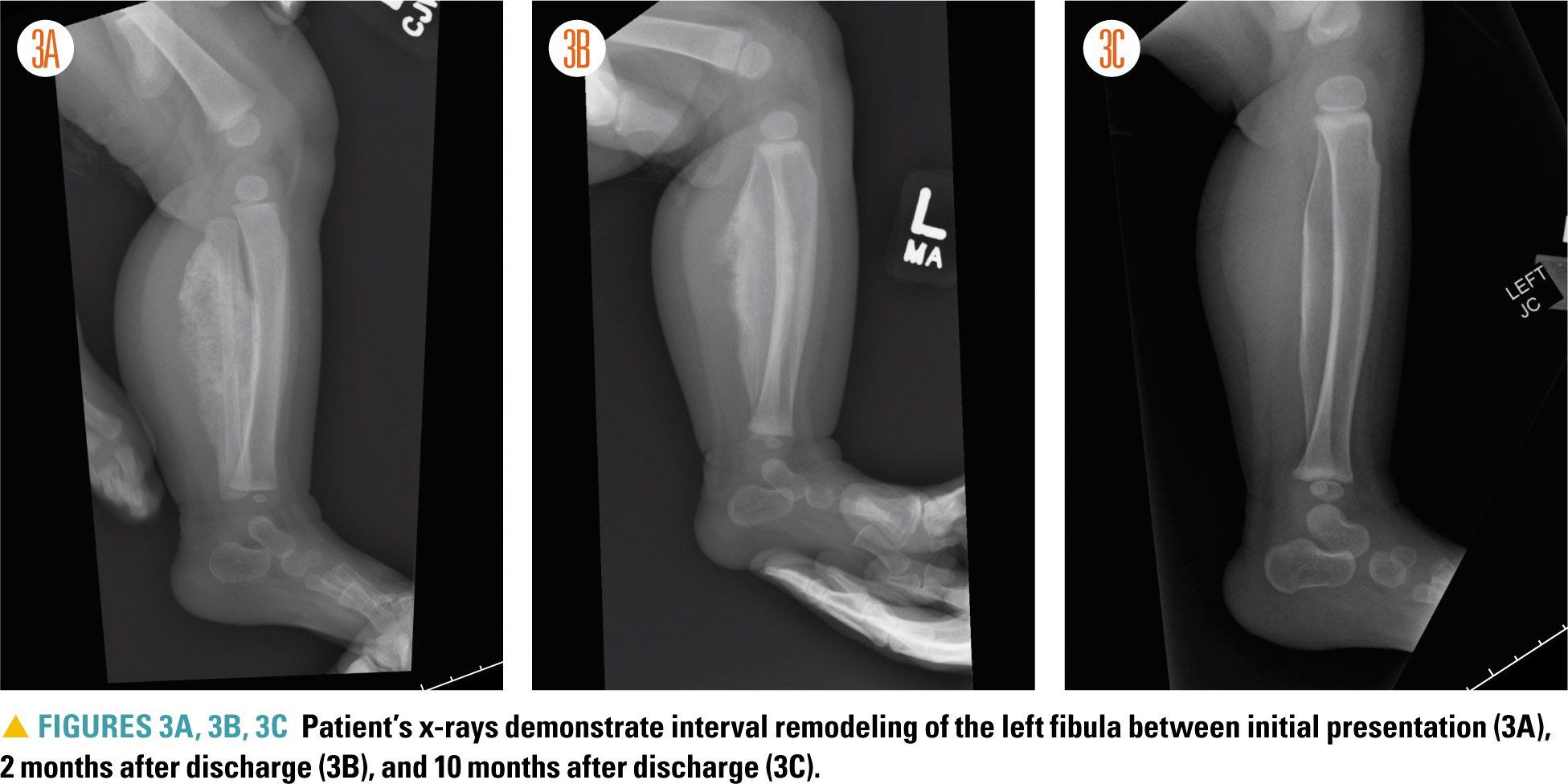
Figures 3A, 3B, and 3C
The Case
A 5-month-old previously healthy, full-term female presented to a pediatric emergency department (ED) with 2 weeks of left leg swelling. Her parents denied any history of trauma, pain, fevers, weight loss, and easy bruising or bleeding, and family history was negative for cancer. The patient had been feeding and eliminating well. The parents said that x-rays of the patient’s left leg were obtained at a different ED soon after they had noted the sudden-onset swelling. At the time they were told that the x-rays were abnormal, and they decided to report to the pediatric ED because authorization for an outpatient magnetic resonance imaging (MRI) study ordered by the patient’s primary pediatrician was taking too long.
Physical examination
In the pediatric ED, the patient was well appearing. Her temperature was 98.42°F; heart rate was 164 beats per minute; respiratory rate was 28 breaths per minute; blood pressure was 127/68 mm Hg (although she was moving while the measurement was obtained); and her oxygen saturation level was 99% on room air. She weighed 6.8 kg.
Physical examination showed significant swelling below the patient’s left knee without erythema, warmth, or apparent tenderness. The infant had intact pulses, no lymphadenopathy, no hepatosplenomegaly, no rash, and no swelling noted elsewhere. She moved all extremities equally with normal range of motion and strength.
Laboratory studies and imaging
Laboratory studies revealed an elevated white blood cell count (20.15 K/uL) as well as an elevated platelet count (722 K/uL) and elevated inflammatory markers (C-reactive protein, 5.0 mg/dL; erythrocyte sedimentation rate, 44 mm/h). A comprehensive chemistry panel, lactate dehydrogenase, and uric acid were unremarkable. Additionally, x-rays of the patient’s left lower extremity and chest were obtained, which preliminarily showed marked periosteal reaction involving the left tibia and fibula and multiple ribs on the left (Figures 1A and 1B).
Differential diagnosis
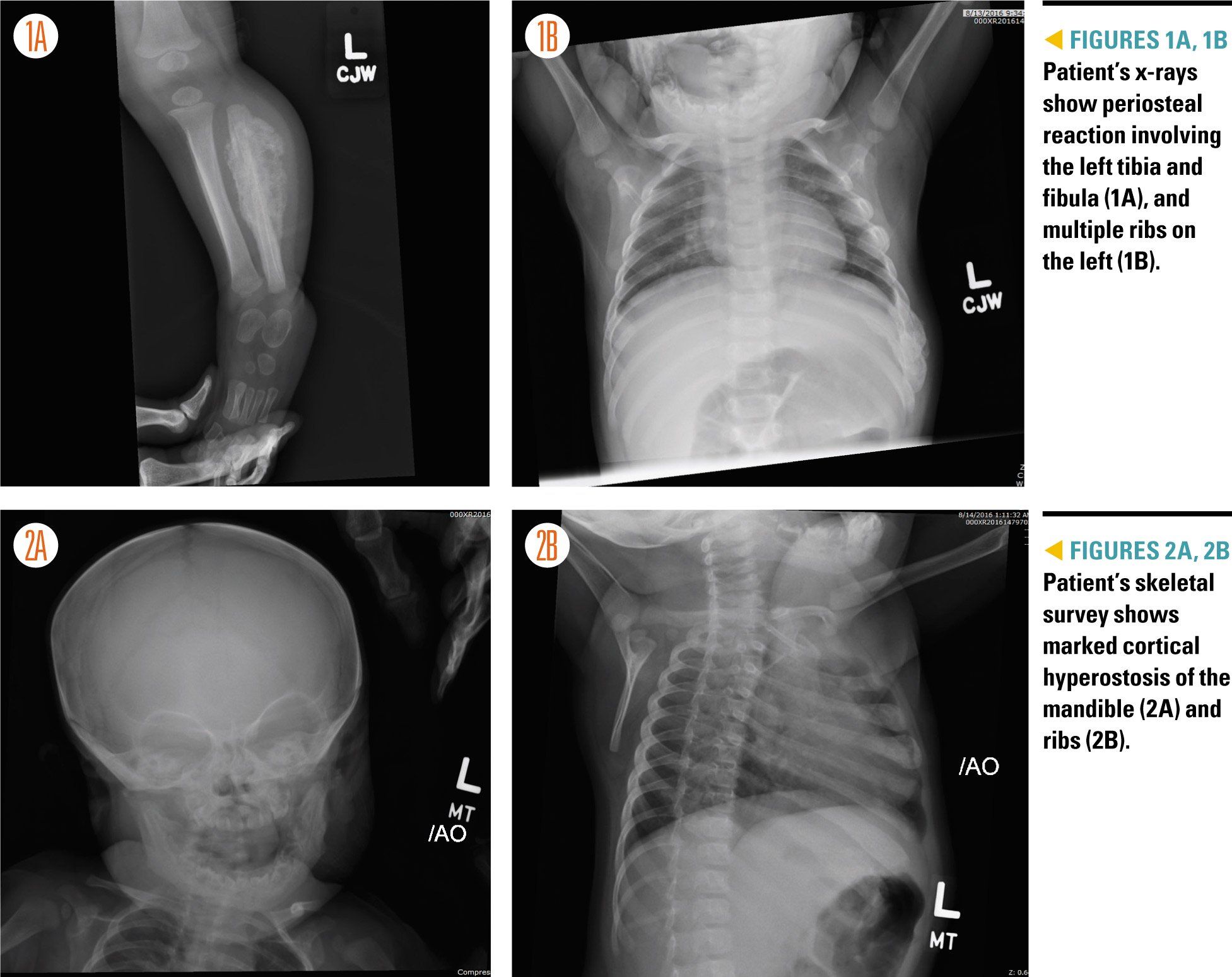
The differential diagnosis in the ED included trauma, osteomyelitis, and malignancy (Table). Other conditions on the differential for unilateral lower extremity soft tissue swelling can be ruled out by the patient’s age of presentation, other associated symptoms, and pattern of bone involvement.1Primary bone malignancies such as osteosarcoma and Ewing sarcoma are very rare in infants. Benign bone and soft tissue tumors such as chondroblastomas and osteoid osteomas do not typically present with marked periosteal reaction in multiple areas. Vitamin deficiencies such as scurvy and rickets are not self-limiting and typically present with other clinical findings (eg, petechiae, corkscrew hairs, and gum disease in scurvy). Exposure to medications such as prostaglandin E (eg, in infants with cyanotic congenital heart disease) and vitamin A can be obtained on history.2,3 Prostaglandin E is thought to promote osteogenesis by decreasing bone resorption and vitamin A is thought to regulate osteoblasts and osteoclasts. Accidental and nonaccidental trauma should be ruled out with careful history taking and scrutiny of the distribution of bone involvement.4 Other important diagnoses to include in the differential are infection (eg, osteomyelitis), endocrine disorders, and rare metabolic disorders.
Consultations and additional studies
The Orthopedic Surgery and Oncology services were consulted due to a strong concern for malignancy. A skeletal survey was obtained (Figures 2A and 2B), and the patient was admitted to the Oncology Service for further management, with the tentative plan by Oncology to obtain an MRI of the left leg and a computed tomography (CT) scan of the chest the following morning.
Upon further review of the patient’s x-rays and skeletal survey, multiple specialists-including pediatric radiologists-agreed that the imaging showed marked cortical hyperostosis of the mandible, ribs, and left tibia and fibula-diagnostic of Caffey disease.
Discussion
Caffey disease (also known as infantile cortical hyperostosis, Caffey-Silverman syndrome, or Smyth syndrome) is a rare, self-limiting condition that should be included in the differential diagnosis of soft tissue swelling in infants. It presents in approximately 3 per 1000 infants prior to 6 months of age, although this is thought to be an underestimation due to underdiagnosis. The condition affects males and females equally and has no ethnic predilection.5 The average age of onset is 9 weeks.6 It is usually characterized by irritability, soft tissue swelling, and cortical bone thickening; however, irritability was not seen in this patient.7,8
Delayed diagnosis may occur because the disorder can present with nonspecific symptoms such as fever, fussiness, and decreased appetite, and therefore can mimic other diagnoses, such as malignancy, trauma, and infection. Swelling usually occurs suddenly and may be red, firm, and/or tender.7 Laboratory tests may reveal leukocytosis, thrombocytosis, and/or elevated inflammatory markers, all of which were noted in this patient.
The etiology of Caffey disease is unclear, but it is thought to be a type I collagenopathy. Both sporadic and familial forms occur. The mandible is the most likely affected bone in sporadic cases, whereas the tibia is most likely affected in familial cases. Autosomal dominant and autosomal recessive patterns of inheritance have been reported by reviewing pedigrees of unrelated families.9
In families with autosomal dominant inheritance, a missense mutation has been found in COL1A1, which codes for the alpha 1 chain of type I collagen.6,10 Interestingly, other reported mutations that affect the synthesis of type I collagen include osteogenesis imperfecta (OI) and Ehlers-Danlos syndrome. It is unclear why a missense mutation would stimulate hyperostosis, and it is also unclear why this mutation would affect individuals at a specific age.
The autosomal recessive pattern is distinct from the classic presentation of Caffey disease in that it is associated with prenatal onset and has a much more severe course (eg, angulations and shortness of long bones, polyhydramnios, and fetal hydrops, in addition to marked hyperostosis) and a poorer prognosis.11
With good clinical suspicion and multidisciplinary input, early identification of Caffey disease can prevent unnecessary workup and invasive procedures. Diagnosis is made by physical exam and radiographic findings. Although imaging may be variable during the clinical course, the classic finding is cortical diaphyseal periosteal new bone formation. Usually x-ray suffices to clinch the diagnosis; however, MRI could add additional value in diagnosis if infection or malignancy is high on the differential or if there is a question of subperiosteal hemorrhage.12
Because Caffey disease doesn’t affect epiphyses or metaphyses, growth potential should not be affected. The pattern of bone involvement is usually multifocal and asymmetric.5 Involved bones typically include the mandible, clavicles, ribs, and long bones. The involvement of the mandible-the most frequently affected bone-is usually pathognomonic and affected in 70% to 90% of cases.8 With mandibular involvement, infants may refuse to eat, leading to failure to thrive.
Caffey disease usually resolves within 6 to 12 months. Treatment typically consists of nonsteroidal anti-inflammatory drugs (NSAIDs) as needed. The use of steroids has been proposed to accelerate bone remodeling and may be trialed if there is a poor response to NSAIDs. In rare cases, Caffey disease can cause enough long-term bony deformity (eg, excessive hyperostosis leading to fusion of adjacent bones) that surgical correction is needed. Other complications include exophthalmos, pleuritis, and recurrence of disease.7,8
Patient outcome
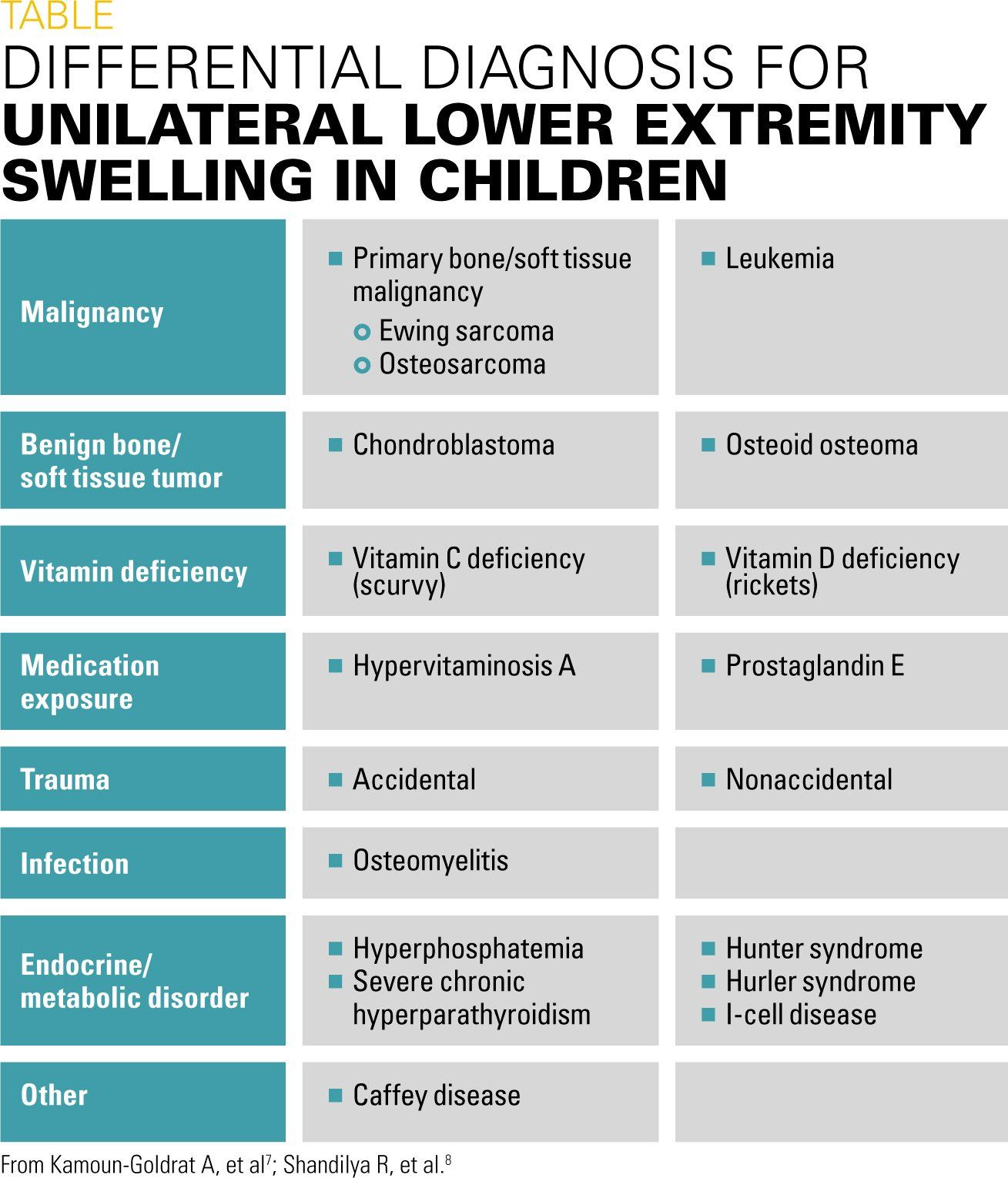
For this patient, no additional imaging was obtained, and she did not require any medical or surgical intervention. She was discharged home with plans to follow up with Orthopedic Surgery. Serial x-rays obtained during outpatient clinic visits with Orthopedic Surgery demonstrated interval bone remodeling (Figures 3A, 3B, 3C), with marked improvement and minimal residual deformity noted 10 months after discharge.
References:
1. Menashe SJ, Iyer RS, Parisi MT, Otto RK, Stanescu AL. Pediatric chest radiographs: common and less common errors. AJR Am J Roentgenol. 2016:1-9.
2. Velaphi S, Cilliers A, Beckh-Arnold E, Mokhachane M, Mphahlele R, Pettifor J. Cortical hyperostosis in an infant on prolonged prostaglandin infusion: case report and literature review. J Perinatol. 2004;24(4):263-265.
3. Woo K, Emery J, Peabody J. Cortical hyperostosis: a complication of prolonged prostaglandin infusion in infants awaiting cardiac transplantation. Pediatrics. 1994;93(3):417-420.
4. Christian CW, States LJ. Medical mimics of child abuse. AJR Am J Roentgenol. 2017;208(5):982-990.
5. Prior AR, Moldovan O, Azevedo A, Moniz C. Caffey disease in neonatal period: the importance of the family! BMJ Case Rep. 2012;2012:bcr2012006996.
6. Glorieux FH. Caffey disease: an unlikely collagenopathy. J Clin Invest. 2005;115(5):1142-1144.
7. Kamoun-Goldrat A, le Merrer M. Infantile cortical hyperostosis (Caffey disease): a review. J Oral Maxillofac Surg. 2008;66(10):2145-2150.
8. Shandilya R, Gadre KS, Sharma J, Joshi P. Infantile cortical hyperostosis (Caffey disease): a case report and review of the literature--where are we after 70 years? J Oral Maxillofac Surg. 2013;71(7):1195-1201.
9. Barba WP, Freriks DJ. The familial occurrence of infantile cortical hyperostosis in utero. J Pediatr. 1953;42(2):141-150.
10. Gensure RC, Makitie O, Barclay C, et al. A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. J Clin Invest. 2005;115(5):1250-1257.
11. Schweiger S, Chaoui R, Tennstedt C, Lehmann K, Mundlos S, Tinschert S. Antenatal onset of cortical hyperostosis (Caffey disease): case report and review. Am J Med Genet A. 2003;120A(4):547-552.
12. Sanders DG, Weijers RE. MRI findings in Caffey's disease. Pediatr Radiol. 1994;24(5):325-327.




Boy presents with fatigue, minimal responsivity, and diffuse muscle weakness
August 7th 2024An 11-year-old boy with a history of asthma and allergic rhinitis presented to the emergency department (ED) with worsening fatigue, minimal responsivity to external stimuli, and diffuse muscle weakness for 2 months.

A 9-year-old boy presents with neck mass
July 3rd 2024A 9-year-old boy was seen for follow-up of a neck mass noted several years earlier. He first presented with this finding at 3 years of age, when during an otherwise unremarkable examination, he became upset, and a protuberant swelling was noted in the anterior aspect of the right side of his neck. What's the diagnosis?

