



Massive splenomegaly in a 6-year-old girl
A 6-year-old female with history of previously resolved iron-deficiency anemia presents to the emergency department (ED) for numerous episodes of nonbloody, nonbilious vomiting and diffuse abdominal pain that began on the day of presentation. She had initially presented to her pediatrician who felt a large left-upper-quadrant abdominal mass and referred her to the ED for further evaluation. She has no associated diarrhea or urinary symptoms. What's the diagnosis?


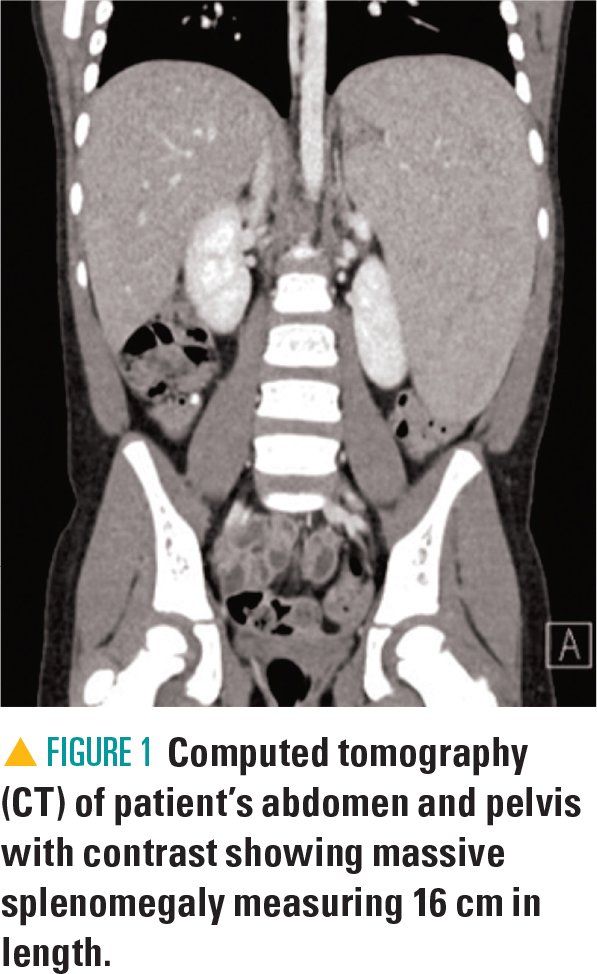
Figure 1


Table


The case
A 6-year-old female with history of previously resolved iron-deficiency anemia presents to the emergency department (ED) for numerous episodes of nonbloody, nonbilious vomiting and diffuse abdominal pain that began on the day of presentation. She had initially presented to her pediatrician who felt a large left-upper-quadrant abdominal mass and referred her to the ED for further evaluation. She has no associated diarrhea or urinary symptoms.
The patient’s mother noticed that her daughter’s abdomen had gradually been looking larger and fuller over the past several days, and today the child began vomiting whenever she ate or drank. Her mother also described a flat, nonpruritic rash that appeared diffusely on her daughter’s body over the preceding days. When asked about other bruises or abnormal bleeding, the mother shared that the child had been having epistaxis bilaterally more frequently and severely than ever before, now occurring most days and without excessive nose-blowing, picking, or recent respiratory illness. The remainder of the patient’s review of systems was negative including fever, household sick contacts, and abdominal trauma.
Evaluation and testing
On initial examination, the patient was noted to be very well appearing. She was smiling, friendly, talkative, and did not appear to be in any distress. Her skin showed diffuse petechiae on the face, arms, legs, abdomen, and back. She had no mucosal lesions. Her abdomen was soft but distended. She had mild tenderness diffusely to palpation but when asked where it hurt the most she pointed to her epigastric region. She had no rebound tenderness or guarding.
The patient was found to have massive splenomegaly measuring approximately 6 cm below the left costal margin without hepatomegaly. The spleen extended toward the pelvis and medially toward midline, palpable just lateral to the umbilicus. The remainder of her physical exam was otherwise unremarkable.
At this time, an abdominal ultrasound was performed to confirm that what was initially described as a right upper-quadrant mass was her enlarged spleen. Abdominal and pelvic computed tomography (CT) was also obtained at this time in the ED to better assess for mass effect of her organomegaly, further characterize the spleen, assess for abdominal lymphadenopathy, and confirm a normal-sized liver (Figure 1). At this time, clinicians presumed that her severe splenomegaly was causing a pseudo-obstructive effect resulting in emesis with oral intake.
Initial bloodwork was obtained including a complete blood count (CBC) and coagulation studies given her diffuse petechiae on exam and history of recurrent epistaxis. Her CBC showed significant pancytopenia with leukopenia to 3.9 K/uL with absolute neutrophil count (ANC) of 1300 K/uL; normocytic anemia with hemoglobin (Hgb) of 10.2 g/dL; hematocrit (Hct) of 29%; however, platelets clumped so were unable to be calculated on this first sample. Her prothrombin time (PT) was elevated to 16.5 sec with an international normalized ratio (INR) of 1.4 sec.
Given her vomiting, electrolytes were obtained using a comprehensive metabolic panel that revealed normal electrolytes, renal function markers, and unremarkable liver function tests. Due to her pancytopenia with neutropenia, there was now concern for a malignancy as cause for her symptoms, so uric acid and lactate dehydrogenase (LDH) were added to her bloodwork, often elevated in leukemia and other malignancies and helpful to trend if concerned for tumor lysis syndrome. At this time, both tests were normal.
The patient was admitted to the hospital with plans to observe overnight. Labs were repeated the next day and showed interval worsening of her pancytopenia: WBCs were 2.0 K/uL; ANC, 800 K/uL; Hgb, 7.8 g/dL; Hct, 23%; and platelets were now calculated at 26 K/uL. At this time, it was decided to include various subspecialists in the patient’s care and assessment of her condition given the broad differential diagnosis. With their guidance, additional tests were performed to ultimately diagnose the patient’s condition.
Differential diagnosis
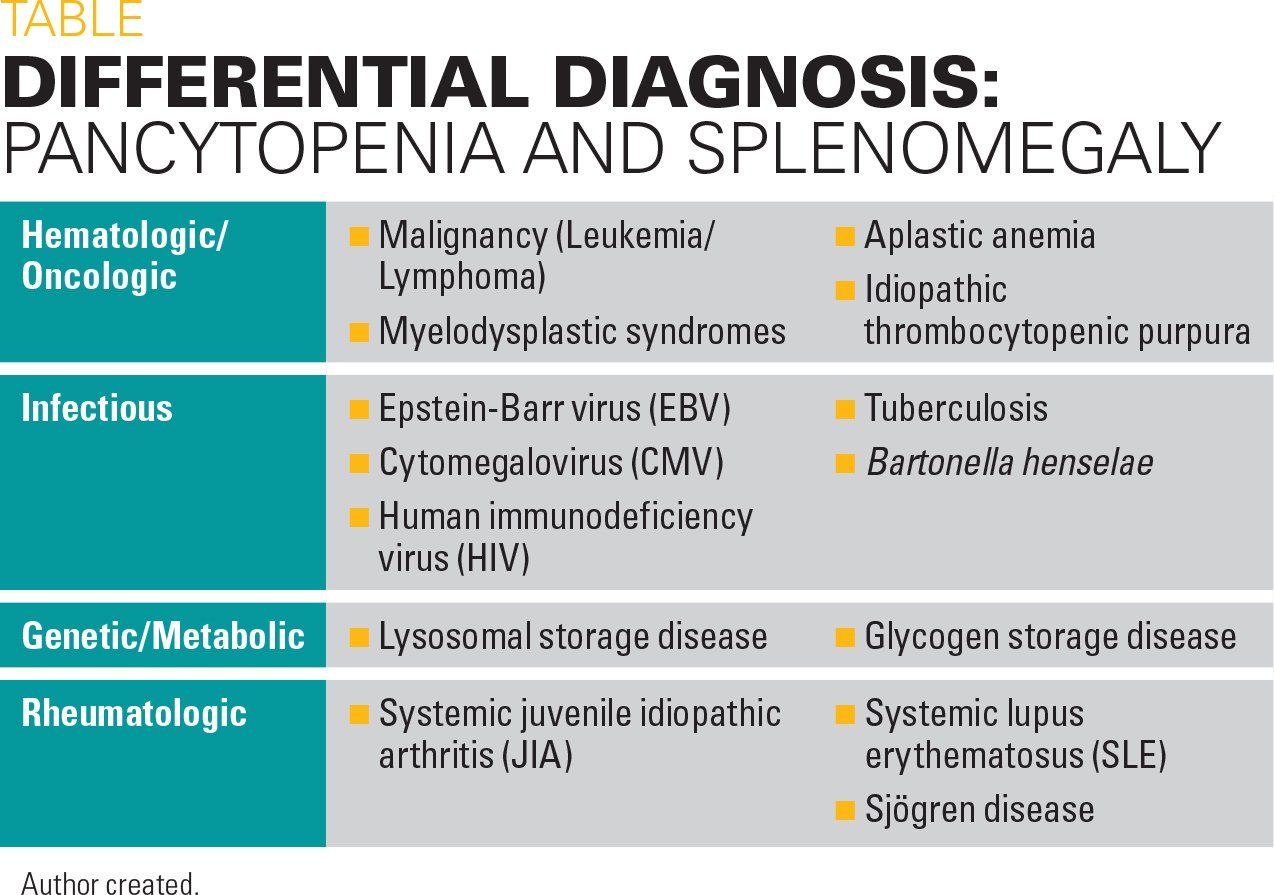
The etiology of pancytopenia in the pediatric population is diverse, ranging from transient bone marrow suppression secondary to acute infection and nutritional deficiencies to hematologic malignancies and bone marrow failure syndromes (Table).
A review study in 2010 estimated that 64% of pediatric pancytopenia is caused by infectious origins, 28% is attributed to primary hematologic/oncologic causes, and the remaining 8% is due to miscellaneous etiologies including metabolic disease.1 For this reason, common infectious causes were considered, including Epstein-Barre virus (EBV), cytomegalovirus (CMV), parvovirus, and human immunodeficiency virus (HIV).2 Testing was performed for these etiologies and ultimately all were negative. Bacterial causes such as Bartonella and tuberculosis were considered less likely at this time given the patient’s low risk of exposure when asked related questions in her history.
Rheumatology was consulted as several conditions including systemic lupus erythematosus (SLE), systemic juvenile idiopathic arthritis (JIA), and Sjögren syndrome can cause splenomegaly, and an antinuclear antibody (ANA) sent at the time of admission showed moderate elevation to 1:320. Additional rheumatologic workup including complement levels and double-stranded DNA (dsDNA) were obtained but anticipated to be unrevealing as the patient lacked other systemic symptoms of these illnesses such as arthritis and skin findings.
Hematology/Oncology was consulted for further assessment of malignancy with primary concern for leukemia over lymphoma or a solid mass given CT findings and the lack of abnormal lymph nodes on this study and by physical exam. Other hematologic conditions such as myelodysplasia or aplastic anemia, which could be secondary to infection, could cause this degree of marrow suppression and given the persistence and worsening of her pancytopenia, there was a question about the utility of a bone marrow aspirate to further assess the appearance of her marrow cells.
Although reassuring for this patient’s overall clinical status to not have laboratory evidence of overt tumor lysis, the findings did little to reassure against a diagnosis of malignancy. With a low white blood cell count, tumor lysis could be absent despite the presence of acute malignancy, as tumor lysis is more likely to occur in a patient with a high tumor burden.3 Furthermore, normal LDH, uric acid, and electrolytes have very little significance in the evaluation of a plausible, nonmalignant underlying diagnosis such as aplastic anemia or myelodysplastic syndromes.
Given the concern for malignancy, bloodwork was sent for flow cytometry and cytogenetics and found to be negative. Her bone marrow suppression persisted despite several days of hospitalization without any meaningful cell recovery. In a patient with persistent pancytopenia in the absence of a clear diagnosis, and particularly in this patient who also presented with splenomegaly, bone marrow evaluation is the gold standard for diagnosis or exclusion of malignancy, regardless of peripheral blood lab results. Additionally, the aim of the bone marrow evaluation is not only to properly assess for evidence of malignant cells, but also to evaluate for other nonmalignant etiologies.4,5 In the case of a patient with persistent pancytopenia, peripheral blood flow cytometry that does not identify any malignant cells would not obviate the need for bone marrow studies, as the clinical picture still suggested some pathology of the bone marrow and therefore further workup was warranted.
At this time, the decision was made to proceed with bone marrow biopsy to assess for both primary marrow production states and infiltrative processes. This led to further discussion of infiltrative processes that also may cause splenomegaly. Abnormal deposition of substances such as proteins or lipids can cause a similar presentation. Thus, genetic metabolic conditions including glycogen and lysosomal storage disorders are included in the differential diagnosis. An angiotensin converting enzyme (ACE) level was obtained to access for this and found to be elevated to 211 u/L.6
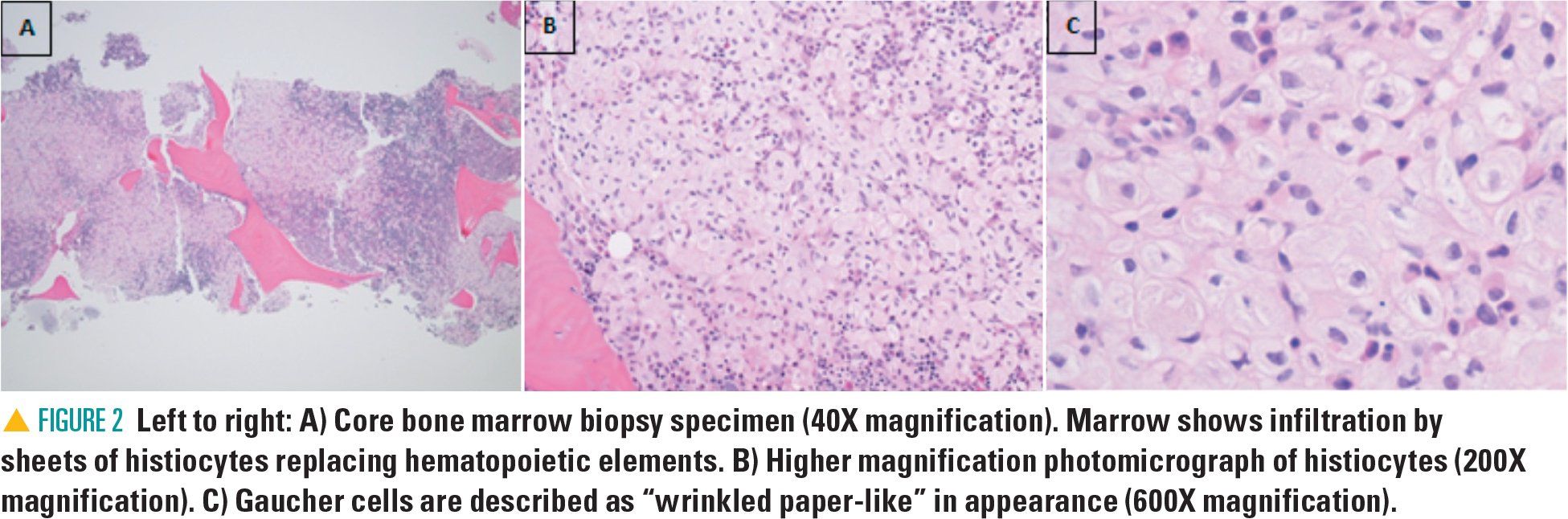
The bone marrow biopsy revealed Gaucher cells, macrophages filled with lipid material that accumulate in Gaucher disease (GD) and that are a primary feature. These cells have a characteristic histologic appearance (Figure 2). However, there are several conditions including hematologic malignancies in which Gaucher-like cells, or “pseudo-Gaucher,” cells have been noted, particularly in circumstances with high cell turnover.7,8 Glucosylceramide is a major component of cell membranes and macrophage glucocerebrosidase activity may become saturated in disorders associated with high cellular turnover. Pseudo-Gaucher cells are benign histiocytes with abundant cytoplasm resembling Gaucher cells on light microscopy.7 However, Gaucher cells have a distinct appearance on electron microscopy that helps to distinguish them from pseudo-Gaucher cells, and this has been suggested to express a different cytokine profile.8 Furthermore, with suspicion of GD based on the noted Gaucher cells, confirmatory testing should be completed to accurately diagnose a patient.
Discussion
The diagnosis of GD should be made by measuring glucocerebrosidase activity in peripheral leukocytes and confirmed via gene mutation analysis of the GBA gene.9 Ideally, it would not be made by finding Gaucher cells on bone marrow biopsy because this study is not necessary to make the diagnosis and is more invasive than enzymatic analysis. In addition to confirming the diagnosis, gene mutation analysis can be of clinical utility, as there is a genotype-phenotype correlation with some mutations. For example, patients homozygous or compound heterozygous for the c.1226A>G allele, as with this patient, have type 1 GD, rather than a neuronopathic type of GD. Type 1 is the most common form and is the non-neuronopathic form of GD.9 It is also the one for which the best therapies are available.
Patient follow-up
Goals of therapy include eliminating or minimizing symptoms, prevention of comorbidities associated with the disease, and improvement in growth and overall quality of life.10 This patient had a port-a-cath placed and was started on enzyme replacement therapy (ERT) with recombinant glucocerebrosidase, given both the severity of her laboratory markers and physical exam findings causing symptomatic disease. This would require every-other-week intravenous (IV) infusions for the foreseeable future, with regular monitoring of labs and imaging.
The patient and her family were counseled not only on the basics of her disease, as it was not known to cause any symptomatic disease in known family members, but also the long-term sequelae and comorbidities of GD; the genetic tests that would be performed; the frequency of additional future testing; and the burden of management with ERT. Her family was counseled further on the autosomal recessive genetic component of her disease and the impact this might have on future family planning.
As both parents were carriers for a pathogenic allele, their future offspring had a 1 in 4 chance of having the disease.9 Additionally, as patients with type 1 GD can have very different phenotypes even with the same genotype, clinicians discussed with the parents the utility of performing genetic testing for the mutations for the patient’s 3 sisters.
Conclusion
Gaucher disease presents similarly to malignancy in many cases, as seen with the patient in this case. Common presenting symptoms include hepatomegaly and/or splenomegaly, bone marrow suppression, bleeding, and bone pain.9,10 It should be included in the differential diagnosis for a patient presenting with symptoms such that clinicians might be considering leukemia as a potential diagnosis.
References:
1. Pine M, Walter AW. Pancytopenia in hospitalized children: a five-year review. J Pediatr Hematol Oncol. 2010;32(5):e192-e194.
2. Rafee Y, Xavier A, Antoine M, Borkin M, Rongkavilit C. An infant with fever, hepatosplenomegaly, and pancytopenia. Pediatr Infect Dis J. 2008;27(6):571.
3. Mirrakhimov AE, Voore P, Khan M, Ali AM. Tumor lysis syndrome: a clinical review. World J Crit Care Med. 2015;4(2):130-138.
4. Desalphine M, Bagga PK, Gupta PK, Kataria AS. To evaluate the role of bone marrow aspiration and bone marrow biopsy in pancytopenia. J Clin Diagn Res. 2014;8(11):FC11-FC15.
5. Memon S, Shaikh S, Nizamani MA. Etiological spectrum of pancytopenia based on bone marrow examination in children. J Coll Physicians Surg Pak. 2008;18(3):163-167.
6. Lieberman J Beutler E. Elevation of serum angiotensin-converting enzyme in Gaucher’s disease. N Engl J Med. 1976;294(26):1442-1444.
7. Carrington PA, Stevens RF, Lendon M. Pseudo-Gaucher cells. J Clin Pathol. 1992;45(4):360.
8. Knox-Macaulay H, Bhusnurmath S, Alwaily A. Pseudo-Gaucher’s cells in association with common acute lymphoblastic leukemia. South Med J. 1997;90(1):69-71.
9. Stirnemann J, Belmatouq N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation, and treatments. Int J Mol Sci. 2017;18(2):E441.
10. Bennet t LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother. 2013;47(9):1182-1193.















Boy presents with fatigue, minimal responsivity, and diffuse muscle weakness
August 7th 2024An 11-year-old boy with a history of asthma and allergic rhinitis presented to the emergency department (ED) with worsening fatigue, minimal responsivity to external stimuli, and diffuse muscle weakness for 2 months.

A 9-year-old boy presents with neck mass
July 3rd 2024A 9-year-old boy was seen for follow-up of a neck mass noted several years earlier. He first presented with this finding at 3 years of age, when during an otherwise unremarkable examination, he became upset, and a protuberant swelling was noted in the anterior aspect of the right side of his neck. What's the diagnosis?

