|Articles|July 1, 2007
- Consultant for Pediatricians Vol 6 No 7
- Volume 6
- Issue 7
Erythropoietic Protoporphyria and Atopic Eczema With Coinfection
Author(s)Kirk Barber, MD, FRCPC
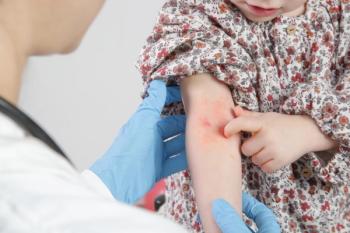
This infant was brought to the emergency department after a week-long exacerbation of her atopic eczema. She had not received any therapy for her eczema or for these erosions.
Advertisement
Case 1:
This young girl has liver failure. Her skin changes confirm the diagnosis and define the cause.
The patient's answer to a single question can replace laboratory diagnostic tests. What would you ask?
Case 2:
This infant was brought to the emergency department after a week-long exacerbation of her atopic eczema. She had not received any therapy for her eczema or for these erosions.
What diagnosis do you suspect?
Case 1: The patient's answer to your question "what happens to your skin when you expose yourself to sunlight?" may avert the need for lab diagnostic tests.
This young woman has erythropoietic protoporphyria (EP). EP is an inherited photodermatosis caused by a deficiency of ferrochelatase--the terminal enzyme in the heme pathway--that results in the accumulation of protoporphyrin in red blood cells, liver, and skin. This produces a severely debilitating lifelong and life-altering photosensitivity. EP is associated with liver failure in up to 4% of those affected.
The typical cutaneous changes of EP are those of scarring of the photo-exposed skin--particularly about the face and hands. The skin takes on a waxy, pebbly, and superficially scarred look that is accentuated over the nose and knuckles. The scars are usually fine and linear, which gives the skin a weather-beaten appearance. Prolonged sun exposure can result in the acute presentation of superficial erosions and crusts on the sun-exposed areas.
The confirmatory laboratory test is the assessment of the erythrocyte protoporphyrin level. This is usually performed in a reference laboratory. In my locale, I call ahead to organize the test. A mild hypochromic microcytic anemia may also be present.
Early recognition of EP can dramatically improve the lives of those affected. Although this disorder is rare (1 in 75,000 to 200,000 is affected), I think every practitioner should be able to make the diagnosis quite easily.
In preparing this case, I returned to an article published in 2006 that reviewed the largest cohort of EP patients ever.1 This is a monumental work that I doubt will ever be reproduced and deserves our respect and attention. Dr Holme of Cardiff, Wales, personally interviewed 223 patients with EP using standardized questions and examination techniques to document each patient's experience with this condition. His findings provide a comprehensive look at EP.
The median age at onset of symptoms was 1 year, but the median age at diagnosis was 12 years. In 34%, the diagnosis was made after the patient's 20th birthday. The presenting symptom in 85% was a burning sensation of sun-exposed skin within a median time of 20 minutes of exposure. Descriptions varied from the dramatic to the very subtle: 95% of patients reported that the onset of their symptoms was not associated with any visible skin changes.
Each affected person seems to have his or her own level of sensitivity to UV light, reflecting the genetic heterogeneity of the enzyme defect. However, when children are exposed to significant amounts of UV light beyond their personal threshold, skin changes will develop; 80% experience swelling within a median time of 6 hours after exposure. Only 20% of children reported any redness of their skin. The severity of the acute changes is related to the duration of the exposure. As noted, photosensitivity is lifelong. Very few children (13%) noted any change over time: symptoms improved in 14 patients and worsened in 11.
The most enlightening discovery from this work is the effect of the various strategies that patients and their physicians used to control the disease. As you can imagine, photoprotection was the most commonly used intervention: 87% of patients wore clothing for protection (long sleeves and pants, hats, and gloves) and 68% used sunscreens (77% applied it once daily).
The standard physician intervention is the prescription of beta-carotene in doses of 25 to 150 mg/d to attain a blood level of 600 to 800 ng/mL. This level needs to be maintained for approximately 3 months before its effectiveness can be assessed. Beta-carotene had been prescribed for 187 of the study's 223 patients, but only 62 patients (28%) were currently using it. Of those who discontinued beta-carotene therapy, 71% cited the orange discoloration of the skin and body fluids as their main objection (carotenemia).
The systemic complication of most concern is protoporphyric liver disease, which occurs in up to 10% of patients with abnormal liver function tests: 5% experience liver failure. In Dr Holme's group, liver failure developed in only 2 patients (1%); 18 patients (8%) had cholelithiasis.
Holme's article can be critiqued from many perspectives, but the wealth of knowledge that it presents puts our diagnostic and treatment efforts into perspective. First, physicians can improve the lives of affected children with early diagnosis: the key is listening closely to the history, because there is little else that presents in this manner. Second, lifelong photoprotection yields the greatest compliance and improvement in symptoms. This is best accomplished by clothing selection and alterations in lifestyle. Also, the development of sunscreens that extend protection into the visible light range (400 to 410 nm) and are more cosmetically acceptable have provided a significant benefit.
The long-term benefits of beta-carotene seem doubtful; two thirds of those who took the medication did not see significant benefit to warrant continuing it. Paradoxically, UVB-311 phototherapy to "harden" the skin has been a successful novel treatment strategy. So has the adjunctive use of antihistamines for the itching and cholestyramine and chenodeoxycholic acid for liver disease.
Two precautions need to be noted. Anemia is unlikely to be the result of iron deficiency. Thus, iron supplementation should only be given if deficiency is documented because it may exacerbate the porphyria. Also, if a patient with EP requires surgery, remember that all tissues will probably accumulate protoporphyrins. The internal organs are likely to be photosensitive to visible light. Therefore, light filters in the operating room should be employed.
1. Holme SA, Anstey AV, Finlay AY, et al. Erythropoietic protoporphyria in the UK: clinical features and effect on quality of life. Br J Dermatol. 2006;155:574-581.
Case 2: The diagnosis is atopic eczema with bacterial and viral coinfection.
Many atopic children suffer from their eczema during the summer. The majority experience an exacerbation related to an irritant reaction to their sweat and many seem to tolerate grasses on the playgrounds poorly. Also, many people see a recurrence of their herpes labialis during the summer. This combination is a potential problem for atopic children.
You may remember the young boy pictured in my September 2004 article (A). Primary herpes blisters had developed on his cheek after an affectionate kiss from his mother. I continue to see parents who have no idea that "cold sores" are infectious, so I urge you to address this issue with parents whenever you can.
The hands you see here are those of an atopic infant girl. Her 5-year-old sibling recently had a "cold sore." Her clinical presentation showed a flare of atopic eczema. But note the nature of the erosions. The left hand shows diffuse monomorphic erosions over the dorsal aspect, which suggests an infection (B). The monomorphic nature is characteristic of herpes simplex virus infection of atopic skin. The erosions on her right hand have coalesced and have increased in depth, which suggests a concomitant bacterial infection (C). The presence of herpes simplex virus on atopic skin is often associated with Staphylococcus aureus infection. Order cultures for both organisms when you are presented with these acute "flares" of atopic eczema.
Cultures of the lesions grew both herpes simplex virus and S aureus. I hospitalized the patient for aggressive therapy for her eczema and for intravenous antiviral and antibiotic therapy. Despite this therapy, however, contractures developed on her right hand secondary to the scarring.
Herpes simplex virus infections of atopic skin require urgent and careful attention. I recommend systemic antiviral therapy in virtually all instances. Cultures should also be ordered for concomitant bacterial infection to avoid the consequences of scarring. *
References:
REFERENCE:
FOR MORE INFORMATION:
Erythropoietic protoporphyria. Patient information and leaflets. British Association of Dermatologists; March 2005. Available at: http://www.bad.org.uk/public/ leaflets/erythropoietic.asp. Accessed June 18, 2007.
Articles in this issue
about 19 years ago
Vitamin D Deficiency Ricketsabout 19 years ago
Herpes Simplex Dermatitisabout 19 years ago
The Core of the Matterabout 19 years ago
Typhoid and Malaria: Now in Your Waiting Roomabout 19 years ago
The Hidden Dangers of Fashionabout 19 years ago
Gratitudeabout 19 years ago
Infant With Facial Anomaliesabout 19 years ago
Epidermal Inclusion Cystsabout 19 years ago
Causes of Black Tongueabout 19 years ago
Group A -Hemolytic Streptococcal VulvovaginitisAdvertisement
Related Content
Advertisement


Advertisement
Advertisement
Trending on Contemporary Pediatrics
1
Family history alone may not prompt type 1 diabetes screening, survey finds
2
Study finds no association between neonatal circumcision and autism diagnosis
3
Chronic skin disorders linked to psychological distress and poorer quality of life in children
4
Severity Scoring and First-Line Topical Therapy for Pediatric AD
5