|Articles|August 31, 2008
Pediatric Vasculitic Syndromes:
A 7-year-old boy presented to his primary care pediatrician with a 24-hour history of vomiting, abdominal pain, and low-grade fever. The child appeared stable. A viral illness was diagnosed. The child was sent home, and his parents were advised to give him adequate fluids as well as acetaminophen as needed for fever.
Advertisement
A 7-year-old boy presented to his primary care pediatrician with a 24-hour history of vomiting, abdominal pain, and low-grade fever. The child appeared stable. A viral illness was diagnosed. The child was sent home, and his parents were advised to give him adequate fluids as well as acetaminophen as needed for fever.
The patient returned the next day with persistent vomiting, abdominal pain, and bloody diarrhea. Physical examination was notable for periumbilical tenderness.
Abdominal CT scan showed thickened small-bowel loops with vascular engorgement and mesenteric edema. These findings were interpreted as consistent with an infectious process. The complete blood cell count and results of a basic metabolic panel were unremarkable. Blood and stool cultures were negative. The child's home environment was significant for pet turtles, which raised the possibility of salmonella infection.
Infectious gastroenteritis was diagnosed, and the patient was hospitalized for intravenous hydration and observation. Colicky abdominal pain and bloody diarrhea persisted. Although renal function was normal, the urinary sediment was significant for proteinuria and microscopic hematuria.
On hospital day 3, purpuric lesions appeared on the patient's lower extremities, and a diagnosis of Henoch- Schnlein purpura (HSP) was made. The patient continued to have severe pain and bloody stools. Ultrasonography of the abdomen showed no evidence of intussusception. Corticosteroid therapy was started to help manage the severe abdominal symptoms.
The patient's abdominal complaints resolved after a short course of corticosteroids, and his urinary sediment normalized within 6 weeks.
HENOCH-SCHNLEIN PURPURA: AN OVERVIEW
The complexity of the pediatric vasculitic syndromes, their varied presentations, and their uncertain causes make these illnesses a diagnostic challenge. Vasculitis-inflammation directed at blood vessel walls-can affect every organ.1 Initial presentations vary significantly and the tendency toward overlap among these syndromes complicates the clinical picture and often delays diagnosis.
Although pediatric vasculitides probably represent a spectrum of disease, there are instances in which it is important to distinguish among them. Treatment and, ultimately, prognosis may differ.
HSP is the most common of the pediatric vasculitides.1-3 It is a nongranulomatous, small-vessel vasculitis4 characterized by IgA-dominant immune deposits in target organs.3 In most cases, it is a self-limiting disease with a benign course. Rarely, however, significant complications may occur. In complicated cases, the diagnosis must be reassessed and the possibility of an alternate vasculitic syndrome considered.
HSP is predominantly a childhood disease. Children between 3 and 15 years are most often affected; the peak incidence is at age 5 years.2,5 Most epidemiological reports reveal a male predominance, with an average sex ratio of 1.5:1.1,6 The incidence of HSP has been reported as 13.5 to 22.1 per 100,000 children in different countries, which suggests that genetics as well as environmental factors may play a role in its pathogenesis. 2,6-8 There have been clear seasonal outbreaks, most frequently during the winter months, suggesting an infectious trigger.1,2,6
ETIOLOGY AND PATHOGENESIS
The cause of HSP remains uncertain. Although no infectious trigger has yet been confirmed, there is a history of upper respiratory infection in 30% to 50% of cases.2,6 Many infections have been described in association with HSP, including group A streptococcus infection, varicella, parvovirus, hepatitis A and B, tuberculosis, HIV infection, and infection with Bartonella henselae; however, no single causative agent has been identified to date.1,8,9 Vaccinations; foods; insect bites; and some medications, such as thiazides, may also act as precipitating factors.10
The vasculitis of HSP is the result of immunoglobulin-mediated inflammation driven by IgA deposition in small vessels of affected organs. Although there are 2 subclasses of IgA (IgA1 and IgA2), only IgA1 is reportedly involved in HSP.11,12 An unknown antigenic stimulant may be the driving force behind this process, causing elevated IgA levels and activating inflammatory pathways that result in necrotizing vasculitis.11-15
CLINICAL MANIFESTATIONS
The clinical triad that typically characterizes HSP is that of nonthrombocytopenic purpura, arthralgias and/or arthritis, and colicky abdominal pain.2 The order of presentation of these signs and symptoms may vary, but all 3 tend to be present at some point during the course of the disease (Table 1).
Table 1
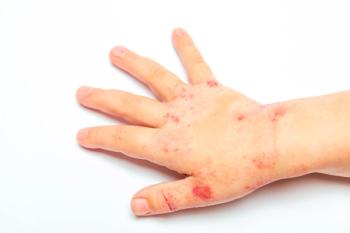
Cutaneous involvement. The presence of palpable purpura is essential for diagnosis of HSP. This requirement is supported by published case series that describe purpura in up to 100% of cases.6,9,16,17 Although there are a few case reports of HSP without the characteristic rash,16,18 its absence increases the likelihood that HSP is not the correct diagnosis.
The rash of HSP is the consequence of immune complex deposition, and it is thus most prominent on dependent or pressure-bearing areas-the lower extremities and buttocks or, in a bedridden patient, the back and extensor surfaces of the arms (Figure). Urticarial wheals or erythematous maculopapules may present first and subsequently transition to purpuric lesions. The lesions may range from very small petechiae to large ecchymoses. Occasionally, lesions may be pruritic, and hemorrhagic bullae have rarely been reported. 2,14 The rash tends to occur in crops and progresses from red to purple to brown. It may last from days to weeks, and it may recur.
Figure – The nonblanching erythematous papules and violaceous flat macules on the legs of this 10-year-old girl were the first clinical manifestation of Henoch-Schnlein purpura. The rash later spread to her arms and face, and edema of the feet and ankles and bilateral ankle pain developed.
Figure
Angioedema secondary to cutaneous vessel damage may be significant and may precede the palpable purpura. It is most prominent over the dorsa of hands and feet and in the periorbital regions; however, in young children it may be seen on the scalp, forehead, and perineum.2,14
Joint involvement. Arthritis and/or arthralgia is the second most common manifestation of HSP, present in up to 75% of cases.6,11 Large joints-in particular the knees and ankles-are most commonly involved. 6,11,14 Joint involvement may precede the rash in up to 25% of cases.9,14 Periarticular swelling, tenderness, and limitation of motion are present without erythema, warmth, or effusion. Joint disease is transient, non-migratory, and although occasionally recurrent, it leaves no permanent deformity.
GI involvement. GI symptoms are seen in 35% to 75% of cases; periumbilical pain and nausea are reported most often.14,19 The symptoms result from edema and ulceration of the bowel mucosa with subsequent intramural hemorrhage.19,20 Hemorrhage is typically confined to the mucosa and submucosa; full-thickness necrosis and perforation are rare.
When GI symptoms precede the rash or are very prominent, an acute abdomen may be suspected and, in some cases, surgical exploration is undertaken.20 Intussusception is seen in 2% to 3% of cases.14 HSP-related intussusception tends to be ileoileal and may be missed on barium enema, making ultrasonography the preferred imaging study.14
Other, rarely seen GI complications include pancreatitis, appendicitis, cholecystitis, protein-losing enteropathy, and hydrops of the gallbladder.2,14,19
Renal and genital involvement. Renal complications of HSP are seen in only 20% to 30% of patients but represent the most common cause of long-term morbidity.2,3 Transient, selflimiting hematuria and proteinuria are most often seen.3 Major complications rarely develop in those patients with hematuria alone, but the presence of proteinuria signals a greater risk of long-term renal morbidity. In a retrospective case study by Calvino and colleagues,17 the best predictor for renal sequelae was the presence of nephrotic syndrome during the first 3 months following the onset of symptoms. Risk factors for nephritis are listed in Table 2.
Table 2
In the initial 6 months of illness, close monitoring of renal status is crucial, particularly in those patients with known risk factors or recurrent disease (because nephritis may present weeks to months after the initial symptoms). Unlike arthritis or GI involvement, nephritis rarely precedes the onset of purpura.6,11 A recent review suggested that even in the face of a normal urinalysis at presentation, late renal manifestations develop in some patients; the authors recommend monitoring of urinalysis for 6 months, particularly in high-risk patients.21
Rarely, renal abnormalities occur more than 6 months after active disease. Persistent nephropathy and progression to end-stage renal disease is rare (occurring in fewer than 1% of cases).14 Renal complications are difficult to treat, and no clear consensus exists as to management. In patients with severe renal involvement with hypertension, other vasculitic processes, such as polyarteritis nodosa, must be considered and appropriately aggressive treatment implemented.
Genital involvement is an uncommon but painful complication of HSP, affecting the scrotal wall, epididymis, testis, testicular appendage, and spermatic cord. Scrotal edema is seen in 2% to 38% of cases and may be severe enough to warrant corticosteroid therapy.13,14 Scrotal symptoms may be the first manifestation of HSP. Differentiation from testicular torsion is crucial, often necessitating surgical exploration. In most cases scrotal symptoms disappear within a month.13
Hematological manifestations. Thrombocytosis, factor VIII and XIII deficiencies, vitamin K deficiency, and hypoprothrombinemia with coagulopathy may complicate HSP. Factor XIII deficiency has been reported in children during the acute phase of the illness, particularly in those with early and predominant abdominal complaints. The level of factor XIII has been suggested as a prognostic indicator, but its true diagnostic value has not yet been clarified.22,23
Pulmonary, internal organ, and deep muscle hemorrhage have been reported.2,14 These are rare complications and, when present, usually require aggressive immunosuppressive medications.24
CNS involvement. Nervous system involvement in HSP is rare, occurring in 2% to 8% of cases.16 The most common symptoms are headaches, mood changes, and seizures. 2,14,16 The combination of vasculitis and hemorrhagic diathesis may lead to a more severe clinical picture that includes cerebral hemorrhage, focal ischemia, or encephalopathy.15,16
DIFFERENTIAL DIAGNOSIS
Although the diagnosis of HSP is ideally made based on the classic triad of clinical manifestations, the 3 elements may not all be clearly present or may not all be present simultaneously. However, these 3 manifestations may be accompanied by a variety of nonspecific symptoms that ultimately evolve into a clear diagnosis. Even when confronted with clinical manifestations typical of HSP, surgical emergencies such as acute abdomen and testicular torsion must occasionally be excluded. When they are needed, imaging and surgical consultation should be obtained promptly (Table 3). Situations in which the diagnosis of HSP is not clear may be further complicated in patients who receive corticosteroid therapy, in whom acute abdominal pain or evolving fever may be masked by medication effects.
Table 3
Occasionally, the classic triad of symptoms is present at disease onset, but the child is more severely affected than would be expected. Exercise extreme caution in this setting, and consider the possibility of an alternate diagnosis. Meningococcemia may present with palpable purpura but is generally accompanied by high fevers and other signs of septicemia. In addition, consider Kawasaki disease and systemic-onset juvenile arthritis, which also present with fever and rash. However, each of the latter illnesses has its own characteristic rash and fever pattern that helps differentiate it from HSP.
Severe abdominal involvement in addition to renal disease and hypertension suggests polyarteritis nodosa. This is a chronic illness of unknown cause that results from the inflammation of small and medium muscular arteries, producing consequent aneurysms.2 Polyarteritis nodosa rarely affects children, but when it does, it is most common in those between 9 and 11 years of age. It affects males and females equally. Clinical manifestations are varied, but up to two-thirds of patients present with nonspecific abdominal pain. Edema of the hands and feet, neurological manifestations, and rash are also seen. The rash ranges from maculopapular to purpuric lesions. Painful subcutaneous nodules are frequently seen.25 Renal disease is usually more severe than that seen in HSP. Renovascular arteritis results in hypertension with proteinuria and hematuria. Diagnosis requires a high index of suspicion and the demonstration of characteristic vascular changes on angiography.
Preceding hepatitis B infection has been strongly correlated with polyarteritis nodosa in adults but is rarely seen in the pediatric population. Antineutrophilic cytoplasmic antibodies may be present but are not diagnostic. Treatment requires corticosteroids and, in severe cases, aggressive immunosuppression with cyclophosphamide.
DIAGNOSIS OF HSP
The European League Against Rheumatism consensus on vasculitis in children recently presented new classification criteria (Table 4). Laboratory testing may be useful when trying to exclude other diseases and for disease monitoring but cannot replace clinical acumen. Levels of acute phase reactants tend to be elevated: thrombocytosis, leukocytosis, an elevated erythrocyte sedimentation rate, and an elevated C-reactive protein level are commonly seen. Results of tests for stool guaiac may be positive in up to 50% of cases.25 Elevated serum IgA levels have been described in up to half of patients with HSP, but they are a nonspecific finding.16,24 Biopsy is the only confirmatory test; however, this is rarely necessary.
Table 4
Kidney biopsy may help pinpoint the diagnosis in unclear or severe cases in which polyarteritis nodosa is a diagnostic possibility. However, this should be undertaken only with extreme caution if at all, because aneurysms of the renal vasculature may cause significant, even lifethreatening blood loss when biopsied. The characteristic biopsy finding is necrotizing vasculitis affecting small blood vessels, with universal vascular deposition of IgA.2
TREATMENT
Treatment of HSP is supportive. Provide adequate hydration, nutrition, and pain control with mild analgesics and NSAIDs. Assess vital signs frequently, with particular attention to blood pressure. The initial workup should include a complete metabolic panel, a complete blood cell count, urinalysis, and stool examination for occult blood. In active disease, perform weekly urinalysis. If a patient is at high risk for nephritis, monitor him or her for at least 6 months.21,26 Further testing should be performed as indicated clinically.
In patients with severe joint pain, NSAIDs may provide some relief. Corticosteroids effectively treat soft tissue swelling as well as joint, scrotal, and GI involvement.14 A recent randomized, double-blind, placebocontrolled trial of 171 patients with HSP confirmed that early prednisone therapy at a dosage of 1 mg/kg/d for 2 weeks, tapered over 2 weeks, is effective in reducing abdominal and joint symptoms but does not prevent renal involvement.27 Recently, a systematic review of 15 articles assessed the benefit of corticosteroids in the treatment of HSP.28 Results suggest that early treatment with corticosteroids may not only increase the odds of inducing resolution of abdominal pain but may also reduce the odds of persistent renal disease. The authors concluded that the potential benefit of early corticosteroid treatment in HSP outweighs its adverse effects. More studies investigating this matter are needed. At this time, the use of corticosteroids in mild cases remains controversial.
For the rare patient with severe renal or extra-renal involvement, corticosteroid toxicity and dependence are a risk, and the use of an additional immunosuppressive agent, such as cyclophosphamide, cyclosporine, or azathioprine, should be considered. 1,5,6,9 Other treatments that have been used in refractory cases- although they are not currently accepted as proven therapeutic modalities- include intravenous immunoglobulin and factor XIII concentrate.29
PROGNOSIS
The prognosis for patients with uncomplicated HSP is excellent.10 Disease tends to last no more than 6 weeks. However, recurrences may be seen in up to half of affected patients; thus, kidney function must be monitored closely for 6 months.14 Longterm morbidity is rare and is most often a consequence of renal or CNS involvement.
References:
- Kim S, Dedeoglu F. Update on pediatric vasculitis. Cur Opin Pediatr. 2005;17:695-702.
- Cassidy JT, Petty RE. Leukocytoclastic vasculitis. In: Cassidy JT, Petty RE, Laxer RM, Lindsley CB, eds. Textbook of Pediatric Rheumatology. 5th ed. Philadelphia: Elsevier Saunders; 2005:496-501.
- Coppo R, Andrulli S, Amore A, et al. Predictors of outcome in Henoch-Schönlein nephritis in children and adults. Am J Kidney Dis. 2006;47:993-1003.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/ PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006; 65:936-941.
- Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med. 1997;337:1512-1523.
- Trapani S, Micheli A, Grisolia F, et al. Henoch- Schönlein purpura in childhood: epidemiological and clinical analysis of 150 cases over a 5-year period and review of literature. Semin Arthritis Rheum. 2005;35:143-153.
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. 2002; 360:1197-1202.
- Ballinger S. Henoch-Schönlein purpura. Curr Opin Rheumatol. 2003;15:591-594.
- Ting TV, Hashkes PJ. Update on childhood vasculitides. Curr Opin Rheumatol. 2004;16:560-565.
- Leung AKC, Robson WLM. Henoch-Schönlein purpura. Consultant For Pediatricians. 2004;3:134-138.
- Saulsbury FT. Henoch-Schönlein purpura. Curr Opin Rheumatol. 2001;13:35-40.
- Saulsbury FT. Clinical update: Henoch-Schönlein purpura. Lancet. 2007;369:976-978.
- Hara Y, Tajiri T, Matsuura K, Hasegawa A. Acute scrotum caused by Henoch-Schönlein purpura. Int J Urol. 2004;11:578-580.
- Lanzkowsky S, Lanzkowsky L, Lanzkowsky P. Henoch-Schönlein purpura. Pediatr Rev. 1992;13: 130-137.
- Chiaretti A, Caresta E, Piastra M, et al. Cerebral hemorrhage in Henoch-Schönlein syndrome. Childs Nerv Syst. 2002;18:365-367.
- Paolini S, Ciappetta P, Piattella MC, Domenicucci M. Henoch-Schönlein syndrome and cerebellar hemorrhage: report of an adolescent case and literature review. Surg Neurol. 2003;60:339-342.
- Calvino MC, Llorca J, Garcia-Porrua C, et al. Henoch-Schönlein purpura in children from northwestern Spain: a 20-year epidemiologic and clinical study. Medicine (Baltimore). 2001;80:279-290.
- Michel BA, Hunder GG, Bloch DA, Calabrese LH. Hypersensitivity vasculitis and Henoch-Schönlein purpura: a comparison between the 2 disorders. J Rheumatol. 1992:19:721-728.
- Aflaki E. Gastrointestinal manifestations of rheumatologic disorders: review of articles. Shiraz E-Medical J. http://pearl.sums.ac.ir/semj/vol3/ jan2002/rheumato&GI.htm. Accessed November 29, 2007.
- Ha HK, Lee SH, Rha SE, et al. Radiologic features of vasculitis involving the gastrointestinal tract. Radiographics. 2000;20:779-794.
- Narchi H. Risk of long term renal impairment and duration of follow up recommended for Henoch- Schönlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child. 2005; 90:916-920.
- Kawasaki K, Komura H, Nakahara Y, et al. Factor XIII in Henoch-Schönlein purpura with isolated gastrointestinal symptoms. Pediatr Int. 2006;48: 413-415.
- Kaneko K, Fujii S, Shono T, et al. Diagnostic value of plasma factor XIII in Henoch-Schönlein purpura. Pediatr Nephrol. 2004;19:702-703.
- Vats KR, Vats A, Kim Y, et al. Henoch-Schönlein purpura and pulmonary hemorrhage: a report and literature review. Pediatr Nephrol. 1999;13:530-534.
- Miller ML, Pachman LM. Vasculitic syndromes. In: Behrman RE, Kliegman RM, Jenson HB, eds. Nelson Textbook of Pediatrics. 16th ed. Philadelphia: WB Saunders Co; 2000:730-731.
- Saulsbury FT. Henoch-Schönlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore). 1999;78:395-409.
- Ronkainen J, Koskimies O, Ala-Houhala M, et al. Early prednisone therapy in Henoch-Schönlein purpura: a randomized, double blind, and placebocontrolled trial. J Pediatr. 2006;149:241-247.
- Weiss P, Feinstein JA, Luan X, et al. Effects of corticosteroid on Henoch-Schönlein purpura: a systematic review. Pediatrics. 2007;120:1079-1087.
- Shin JI, Lee JS. Severe gastrointestinal vasculitis in Henoch-Schönlein purpura: pathophysiologic mechanisms, the diagnostic value of factor XIII, and therapeutic options. Eur J Pediatr. 2007;166:1199-1200.
Advertisement
Related Content
Advertisement



Advertisement
Advertisement
Trending on Contemporary Pediatrics
1
FDA approves acetaminophen-naproxen sodium combination tablet for ages 12 and older
2
US measles cases surpass 2025 total, hit highest level since 1991
3
Pooled phase 3 data show early, consistent tapinarof cream responses across pediatric age groups in AD
4
Prenatal therapy improves outcomes in fetal renal failure
5