Takeaways
- Hydroxyurea treatment linked to treating sickle cell disease in children.
- Study assesses morbidity in sickle cell disease from newborn screening (1986) to hydroxyurea introduction (2015).
- Hydroxyurea effectiveness: Reduces pain crises, acute chest syndrome, anemia, and transfusion requirements.
- Severe genotype group faces heavy transfusion burden, with 299/100 person/years incidence.
- Prospective neonatal cohort study highlights significant morbidity in sickle cell disease without preventive intensification using hydroxyurea.
According to a recent study, Hydroxyurea treatment is linked with treating sickle cell disease in children.
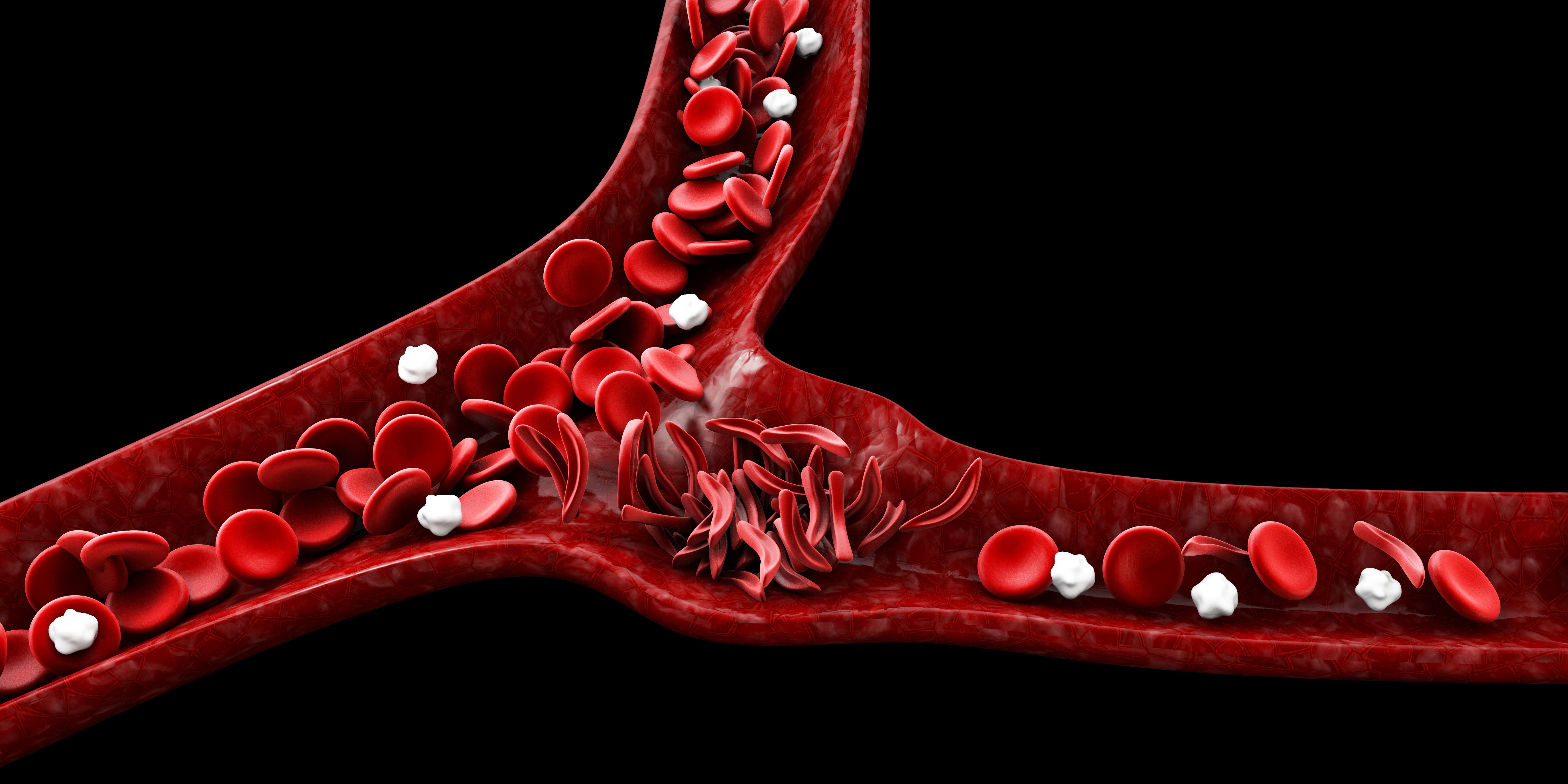
Hydroxyurea, a medicine that helps people with sickle cell disease, keeps blood cells round and flexible so they can do a good job delivering oxygen to the body. The medicine can help certain types of sickle cell diseases, like hemoglobin SS, pain crises, acute chest syndrome, severe anemia needing blood transfusions, and tiny blockages without pain. Hydroxyurea can be taken at just 9 months old.
While newborn screening has reduced early mortality for children with sickle cell disease, few studies have examined the disease difficulties and the transfusion requirement during childhood. After detecting recurring vaso-occlusive crises or acute chest syndrome, a doctor may prescribe hydroxyurea.
The study, led by Alizée Soulié, of the sickle cell disease referral center at Intercommunal Hospital of Creteil in France, evaluated morbidity in children with sickle cell disease from newborn screening (1986) to the introduction of hydroxyurea (2015). The investigators studied a cohort, and the trial lasted from 1 – 20 years.
“This study provides new information about the overall transfusion requirements in [sickle cell disease] as expected, the transfusion burden is particularly heavy in the severe genotype group, with an incidence of transfusion episodes of 299/100 [person/years],” the investigators wrote. “Most transfusion episodes were delivered during chronic [transfusion program] 245/100 [person/years].”
The newborn cohort had 4 genotypes, including SS and Sβ0 thalassemia (n = 289), sickle-hemoglobin C disease (SC n = 65), sickle β+ thalassemia (Sβ+ n = 32), and SDPunjab (SD n = 3). The investigators viewed HbSS, HbSβ0, and HbSDgenotypes as a separate, severe sickle genotype group due to their clinical similarity. There were also HbSD/Sβ0 subgroups.
Ten of the 389 patients died during the follow-up period at a median age of 10.8 years, and 9 deaths occurred because of their sickle cell disease. Everyone but 1 patient had a HbSS disease. One patient with a HbSβ+ disease died at 13 years old due to a pulmonary embolism after a long plane ride.
Participants had a 98.2% chance of surviving at 5 years old (95% CI, 96.6% – 99.3%) and death only occurred in the severe genotype group (97.6%; 95% CI, 95.5% – 99.0%). Meanwhile, participants had a 97.4% chance of surviving at 15 years old, but for HbSS/SD/Sβ0 groups the survival chance was 97.2% (95% CI, 95.0%–98.8%), for HbSC it was 100% (95% CI, 100% –100%), and for HbSβ+ genotype groups it was 90.9% (95% CI, 68.0% – 100%).
“In conclusion, we report here the longest prospective neonatal cohort study to date addressing acute vaso-occlusive, hematological, and extracerebral major organ complications, and providing new data about transfusion requirements in [sickle cell disease] children,” the investigators wrote. “In the absence of preventive intensification with [hydroxyurea], [sickle cell disease] continues to cause substantial morbidity and the use of blood resources remains considerable.”
The investigators wrote how other studies have noted the beneficial effects of hydroxyurea treatment, how the medication reduces pain, dactylitis, acute chest syndrome, hospitalizations, and transfusions. Also, trials indicated that raising the hydroxyurea dose was safe and provided greater clinical benefits than the 20 mg/kg per day dosing. The investigators believe doctors should consider prescribing hydroxyurea earlier with higher doses for children with HbSS/Sβ0 thalassemia.
“We believe that its preventive effects following early introduction and dosage escalation to the [maximal tolerated dose] should alter the course of the disease, and that its substitution for chronic transfusions to prevent primary stroke should decrease overall blood requirements,” the investigators wrote. “Involving individuals with [sickle cell disease] and their families in medical decision making will be critical to increase adherence to treatment.”
Reference:
Soulié A, Kamdem A, Neumann F, et al. Clinical events in a long-term prospective neonatal cohort of children with sickle cell disease: Evidence for a high disease burden without systematic preventive intensification with hydroxyurea [published online ahead of print, 2023 Oct 26]. Am J Hematol. 2023;10.1002/ajh.27142. doi:10.1002/ajh.27142
A version of this article was initially published by our sister publication, HCP Live.