|Articles|January 14, 2020
- Vol 37 No 1
- Volume 37
- Issue 1
Rash triggers joint pain in an 8-year-old girl
Author(s)Vasudha Mahajan, MD
An 8-year-old, previously healthy girl presents to the emergency department (ED) with a rash “that looks likes bruises” and joint pain. The red patchy rash is not painful and not pruritic. What's the diagnosis?
Advertisement
The case
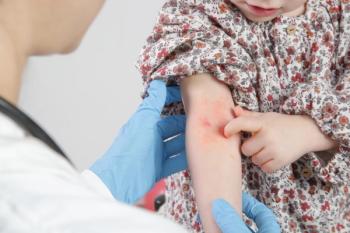
An 8-year-old, previously healthy girl presents to the emergency department (ED) with a rash “that looks likes bruises” and joint pain (Figure). Her mother reports that the rash started over her daughter’s lower legs a week earlier and has since spread to her thighs and buttocks. The red patchy rash is not painful and not pruritic. The girl denies new exposures to food or topical products, recent travel, camping, or recent injury.
History and examination
There is no previous history of easy bleeding or bruising. Both patient and mother deny abuse. No other family members have a similar rash. The patient also reports a 2-day history of new onset left knee and left ankle pain associated with knee swelling, which has since spontaneously resolved. No recent trauma is noted.
The patient denies swelling of her hands or shoulders. She denies chest pain, abdominal pain, dysuria, or hematuria. A week prior to the rash, she was evaluated for fever and sore throat and tested negative for streptococcal infection.
On exam, the child is well appearing, alert, and hydrated. Her weight is 87 lb (98th percentile); temperature is 98.5°F; pulse is 98; respiratory rate is 24 breaths/min; blood pressure is 110/60 mm Hg; and pulse oximetry is 100% on room air.
Her physical exam is negative for conjunctivitis, oral ulcers, or lymphadenopathy. Respiratory and cardiovascular exams are within normal limits. Abdominal exam is negative for tenderness on palpation without guarding or rigidity, and bowel sounds are normal. No hepatosplenomegaly is palpated.
The ankle joints are tender on palpation over the lateral and medial malleolus without any swelling, erythema, deformity, or restriction of motion. The knee and hip joints are normal. Neurologic exam is normal without any focal neurologic defects identified.
Her skin exam is positive for palpable purpuric rash that is nonblanchable and nontender (Figure). She also has an interspersed petechial rash over the lower extremity that extends from the ankles to the thighs, lower abdomen, and buttocks. The soles of her feet are not involved.
Laboratory testing
Initial blood work revealed a complete blood count (CBC) with a slightly elevated white blood cell (WBC) count of 10.5 X 109 /L (3.40-9.5 X 109/L) with a normal differential; hemoglobin, 12.9 g/dL (12-14 g/dL); platelet count of 481 X 10 9/L (150-450 X 109/L). A complete metabolic panel showed normal liver function tests and a normal urea/creatinine ratio. Urinalysis was negative for protein, blood, or leukocytes. Serum antinuclear antibody (ANA) test was negative.
Differential diagnosis
The differential diagnosis for rash with joint pain in children is broad and includes both infectious and noninfectious causes (Table). A detailed history with pertinent positives and negatives and a thorough exam is helpful in making the diagnosis. Some of the common differentials are discussed below.
SYSTEMIC LUPUS ARTHRITIS
Systemic lupus arthritis (SLE)1 is a multisystem autoimmune condition caused by inflammation of the blood vessels and connective tissue. Because this condition can involve multiple systems of the body and occurs in episodic flares, the periodic constellation of symptoms can make it hard to diagnose.
Generalized symptoms such as fever, weight loss, lymphadenopathy, and hepatosplenomegaly along with the classic malar rash and nonerosive symmetric arthritis should raise the suspicion for SLE. Laboratory workup is usually positive for cytopenias, transaminitis, and elevated inflammatory markers. A positive ANA titer is a very sensitive marker but not specific, and in the event that it is positive, follow-up with specific anti–double-stranded DNA (anti-ds-DNA) antibody and anti-smith (Sm) antibody should be done to differentiate SLE from other connective tissue and vascular disorders.
The patient in this case was well appearing without systemic symptoms and had both a normal blood count and a negative ANA, which made SLE less likely.
LYME DISEASE
With Lyme disease,2 the pathognomic of the early localized stage (most common presentation within the first 1 to 4 weeks after a tick bite) is the erythema migrans (EM) rash, which appears as a “target-like lesion” or “bull’s-eye appearance” and can be found on the abdomen, axilla, inguinal, or popliteal areas. This is associated with systemic signs such as fever, arthralgia, and headaches.
Laboratory studies might show leukopenia or leukocytosis, elevated inflammatory markers such erythrocyte sedimentation rate (ESR), and liver function abnormalities. Early disseminated disease secondary to hematogenous spread of the bacteria presents as multiple EM, neurologic involvement including facial nerve palsy, or carditis. Lyme arthritis is the main symptom of late disseminated disease, presenting months to years after the tick bite. Lyme disease can be monoarticular or oligoarticular with small effusions and absence of fever.
No exposure to the tick and absence of the characteristic rash and laboratory abnormalities made this diagnosis less likely in this patient.
RHEUMATIC FEVER
Acute rheumatic fever (ARF)3 is one of the causes of primary acquired heart diseases due to an inflammatory reaction after a streptococcal infection. It is a clinical syndrome that includes criteria as outlined in the Jones criteria.3 Laboratory evidence of a preceding group A streptococcal infection is mandatory for the diagnosis. The rash associated with ARF is erythema marginatum, which is a macular blanching rash characterized by central clearing and found mostly on the trunk and proximal extremities. Migratory polyarthritis and carditis are the most common presenting symptoms.
The rash in this patient did not match the description of the classic rash seen with ARF and her rapid strep test was negative. Since the suspicion for ARF was low based on clinical findings, a throat culture was not ordered.
IDIOPATHIC THROMBOCYTOPENIC PURPURA
Idiopathic thrombocytopenic purpura (ITP)4 is an autoimmune condition resulting in increased platelet destruction secondary to antiplatelet antibodies. It is the most common cause of isolated thrombocytopenia in otherwise well children. It can manifest as sudden onset bruising, petechiae, or mucosal hemorrhage, usually after a viral upper respiratory infection and rarely after live vaccinations. On physical exam, lymphadenopathy might be present secondary to the viral infection, but other systemic signs are absent. Isolated low platelet count below 50 X 103/μL with an elevated mean platelet volume and a normal hemoglobin and total white cell count is the only significant laboratory finding. Most cases are self-limiting within 1 to 4 weeks and treatment with oral steroids, intravenous immunoglobulin (IVIG), or anti-D IG is reserved for children with severe hemorrhage or platelet count less than 20 X 103/μL.
Normal platelet count ruled out ITP in this patient.
HENOCH-SCHONLEIN PURPURA
Henoch-Schonlein purpura (HSP) is an immune-mediated, small-cell vasculitis and can mimic different conditions depending on the system involved: Abdominal pain secondary to gastrointestinal (GI) involvement can present as acute abdomen; joint involvement may mimic juvenile idiopathic arthritis (JIA), rheumatic arthritis, or gonococcal arthritis; and the rash can look similar to ITP, rickettsial diseases, sepsis, or disseminated intravascular coagulation. Laboratory tests are nonspecific for HSP, and more useful is ruling out other potential causes of the presenting symptoms.
Given the characteristic purpuric rash in this otherwise well-appearing patient, along with laboratory findings ruling out the other potential causes, a diagnosis of HSP was made.
Discussion
Henoch-Schonlein purpura is the most common systemic small-vessel vasculitis in children.5 The mean age of presentation is 6 to 10 years, and the incidence is known to be equal in boys and girls. The disease is more common in autumn and spring.
ETIOLOGY AND PATHOGENESIS
The classical pathogenic feature of HSP is deposition of IgA immune complexes in the vessel walls of the kidney and other affected organs. These in turn activate the complement pathway that triggers the inflammatory cascade leading to the clinical picture discussed. The most common trigger for HSP is known to be upper respiratory infection.5 Several other viruses and bacteria including but not limited to influenza, parainfluenza, Epstein-Barr virus (EBV) and Streptococcus have been associated with HSP.
There have been associations of certain vaccinations such as influenza, meningococcal, measles/mumps/rubella (MMR), and pneumococcal as well as certain drugs with the onset of HSP. However, these associations have not been proven to be linked to causality, and in most cases such associations have been presumed coincidental.
CLINICAL FEATURES AND COMPLICATIONS
The diagnosis of HSP is clinical and based on palpable purpura along with one of the following features:
1. Diffuse abdominal pain;
2. Arthralgia or acute arthritis;
3. Renal involvement with proteinuria or hematuria; or
4. Renal biopsy with predominant IgA deposition with leukoclastic vasculitis.
Skin rash in HSP has been described as petechial or palpable purpura. The rash is symmetrical, nontender, and most commonly observed on the extensor surfaces of the dependent portions of the lower extremities and the buttocks. Involvement of the abdomen and face have been described in rare cases.6 Hemorrhagic bullae have been reported in only 2% of the affected children and are not related to the severity or the progression to renal failure.7 In some cases, macular and urticarial rashes have been reported prior to the development of the purpuric rash.6
Abdominal pain is the most common (~75%) associated symptom secondary to visceral purpura causing bowel edema and hemorrhage. Colicky diffuse pain, which is worse after meals and that may be associated with emesis, hematemesis, or guaiac positive stool, is the most common presentation. Although it is associated with purpura the majority of the time, abdominal pain may precede the classic rash in less than 40% of the cases, thereby making the diagnosis a challenge.8 Intussusception is the most common complication of abdominal involvement, and an ultrasound is the initial diagnostic modality for identifying it. More severe complications such as extensive hemorrhage and bowel wall gangrene are rare.
Arthritis/arthralgia is the presenting symptom in approximately 25% of patients with HSP, however, more than 50% of patients have articular involvement.5,9 It is usually oligoarticular and involves the larger joints of the lower extremities. Even though the pain, swelling, and restricted range of movement can cause significant discomfort, the symptoms resolve spontaneously within a few days to weeks and the response to nonsteroidal anti-inflammatory drugs (NSAIDs) is well documented. There is no longterm deformity or chronic damage to the joints or ligaments.9
Renal involvement is seen in 20% to 50% of the patients diagnosed with pediatric HSP.5,10 The earliest finding is microscopic hematuria with or without proteinuria.5 Children aged older than 4 years, purpura persisting for longer than 1 month, and severe GI bleeding are higher risk factors for developing renal disease. The renal symptoms take longer to manifest in comparison with the joint and abdominal symptoms. Several studies have shown that renal involvement was apparent within 3 months of appearance of the purpura in 97% of the patients and within 4 weeks for 75% of the patients.5,10 Based on this, weekly urinalysis and blood pressure measurements must be performed in the first 3 months after diagnosis. About 12% of the children with HSP end up with chronic renal failure about 3 to 4 years after diagnosis.5
Other clinical features that have been reported but are less common are headaches, intracranial hemorrhage, pulmonary hemorrhage, scrotal hematoma, and orchitis.
DIAGNOSIS
Routine blood work and imaging are not needed to diagnose HSP but may be obtained in atypical presentations in order to rule out other diagnoses. Initial investigations may include a CBC, urea, creatinine, coagulation studies, and urinalysis (UA). If the UA is positive for hematuria or proteinuria, it should be followed up with a protein/creatinine ratio and further laboratory tests to rule out other causes of glomerulonephritis. Moderate leukocytosis with a mild increase in acute phase reactants is commonly seen. Even though HSP by definition requires a normal platelet count, mild thrombocytosis may be seen in children with GI involvement. Elevated IgA levels are seen in 50% of the patients with HSP, however, it is very nonspecific and there is also no co-relation of the IgA levels with the disease severity. Complement levels are normal.
Biopsy remains the most specific diagnostic tool but is rarely indicated. It is only considered if the diagnosis is uncertain or if the patient has progressive renal disease or nephrotic range proteinuria. Leukoclastic vasculitis affecting the small blood vessels along with perivascular infiltration of neutrophils is typical.
Immunofluorescence shows with IgA deposits along with possible C3 deposits seen in the mesangial wall.
MANAGEMENT
The management of HSP is primarily supportive and involves hydration and pain control with NSAIDs, and rarely with opioids. The treatment with NSAIDs has not shown to increase the risk of GI bleeding. However, in patients with renal involvement and on NSAIDs, close monitoring of renal function and blood pressure is of utmost importance.
The use of steroids in patients with HSP is controversial. Whereas some studies have shown that early use of steroids decreases the duration of abdominal pain and prevents GI and renal complications, other studies have shown no difference.11,12 Immunosuppressive treatment and renal biopsy are reserved for patients with nephrotic range proteinuria and progressive renal impairment. An essential part of following patients with HSP is monitoring them for complications. An abdominal ultrasound is required for patients with new onset severe abdominal pain. Any change in mental status should prompt an evaluation for intracranial hemorrhage.
PROGNOSIS
The prognosis is generally excellent with the majority of the children experiencing complete resolution of the symptoms within 2 to 3 weeks. One-third of the patients experience a recurrence, usually within the first 6 months of diagnosis.13 A recurrence is defined as reappearance of the symptoms after resolution of the disease for at least 2 weeks. It is more likely to happen in children with nephritis. The age of the patient has not been shown to have any association with recurrence risk.
MONITORING AND FOLLOW-UP
Close monitoring for children diagnosed with HSP is essential. Serial urinalysis and blood pressure checks should be performed weekly for the first month, every 2 weeks until 3 months, and monthly until 6 months.10 Renal involvement suggested by hypertension or macroscopic/microscopic hematuria or proteinuria should prompt a nephrology referral. The risk of renal involvement decreases significantly after 6 months.13
Patient course
The patient in this case is being followed in clinic weekly for urinalysis and blood pressures, which have thus far been negative for hematuria or proteinuria. Her purpuric rash had resolved at about 2 weeks and she now has petechial rashes over the lower extremities. At 3 weeks postdiagnosis, she started complaining of intermittent abdominal pain after meals, and an ultrasound was done that was negative for intussusception. The patient will continue to be monitored as per protocol.
References:
1. Weiss JE. Pediatric systemic lupus erythematosus: more than a positive antinuclear antibody. Pediatr Rev. 2012;33(2):62-73; quiz 74.
2. Carriveau A, Poole H, Thomas A. Lyme disease. Nurs Clin North Am. 2019;54(2):261-275.
3. Webb RH, Grant C, Harnden A. Acute rheumatic fever. BMJ. 2015;351:h3443.
4. Buchanan GR. Thrombocytopenias during childhood. Pediatr Rev. 2005;26(11):401-409.
5. González LM, Janniger CK, Schwartz RA, Pediatric Henoch Schnolein purpura. Int J Dermatol. 2009:48(11):1157-1165.
6. Trnka P. Henoch-Schönlein purpura in children. J Paediatr Child Health. 2013;49(12):995-1003.
7. Trapani S, Micheli A, Grisolia F, et al. Henoch-Schonlein purpura in childhood: epidemiological and clinical analysis of 150 cases over a 5-year period and review of literature. Semin Arthritis Rheum. 2005:35(3):143-153.
8. Choong CK, Beasley SW. Intra-abdominal manifestations of Henoch-Schönlein purpura. J Paediatr Child Health. 1998;34(5): 405-409.
9. Roache-Robinson P, Hotwagner DT. Henoch-Schonlein purpura (anaphylactoid purpura, HSP). StatPearls [Internet]. Treasure Island, FL: StatPearls Publishing. Available at:
10. Sano H, Izumida M, Shimizo H, Ogawa Y. Risk factors of renal involvement and significant proteinuria in Henoch-Schönlein purpura. Eur J Pediatr. 2002;161(4);196-201.
11. Narchi H. Risk of long-term renal impairment and duration of follow-up recommended for Henoch-Schonlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child. 2005;90(9):916-920.
12. Weiss PF, Feinstein JA, Luan X, Burnham JM, Feudtner C. Effects of corticosteroid on Henoch-Schönlein purpura: a systematic review. Pediatrics. 2007;120(5):1079-1087.
13. Tizard EJ, Hamilton-Ayres MJ. Henoch-Schonlein purpura. Arch Dis Child Educ Pract Ed. 2008;93(1):1-8.
Articles in this issue
over 6 years ago
Special diets and supplements: Do’s and don’ts for childrenover 6 years ago
What’s ruining medicine for pediatricians?over 6 years ago
Fluoride exposure in pregnancy can affect offspring’s IQover 6 years ago
Linear papules appear on a boy’s thumb, wristalmost 7 years ago
How bronchodilators can help assess asthma severityalmost 7 years ago
How racial inequality affects health and healthcareAdvertisement
Related Content
Advertisement




Advertisement
Advertisement
Trending on Contemporary Pediatrics
1
Family history alone may not prompt type 1 diabetes screening, survey finds
2
Severity Scoring and First-Line Topical Therapy for Pediatric AD
3
How to Recognize Early Pediatric AD and Rule Out Cradle Cap
4
Chronic skin disorders linked to psychological distress and poorer quality of life in children
5