



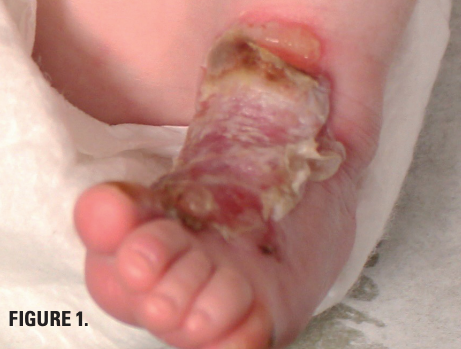
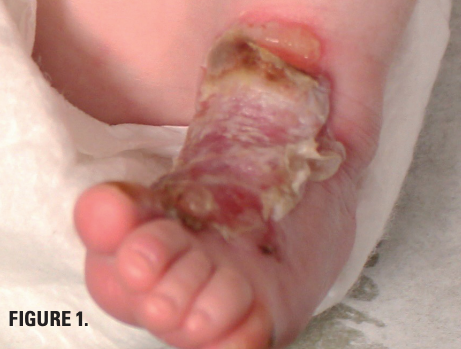
Friction-induced blistering on a child’s feet
You are called to the hospital nursery to evaluate a healthy full-term newborn boy who developed painful flaccid blisters and erosions on the tops of his feet and ankles shortly after birth. His mother had a history of similar recurrent skin lesions that healed with scarring. She also had oral and gastrointestinal tract involvement. What's the diagnosis?
Case
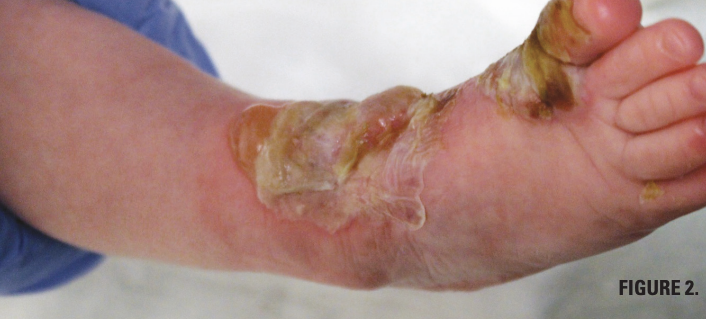
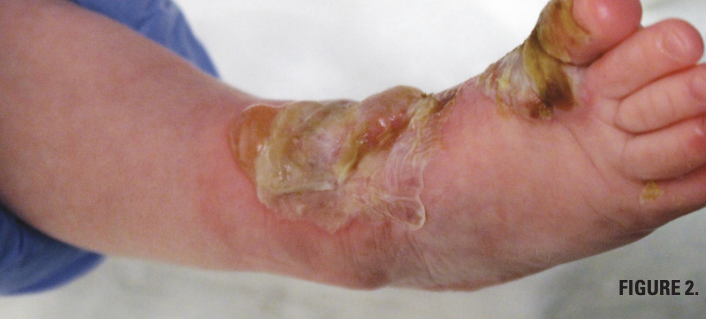
You are called to the hospital nursery to evaluate a healthy full-term newborn boy who developed painful flaccid blisters and erosions on the tops of his feet and ankles shortly after birth (Figures 1 and 2). His mother had a history of similar recurrent skin lesions that healed with scarring. She also had oral and gastrointestinal tract involvement. What's the diagnosis?

Diagnosis:
Dominant Dystrophic Epidermolysis Bullosa (DDEB)
Etiology and clinical findings
Epidermolysis bullosa, a group of rare genetic diseases characterized by skin fragility resulting in the formation of painful bullae and ulcers from minor mechanical trauma, can have devastating medical consequences for patients. Skin lesions can occur on cutaneous and mucosal surfaces and occasionally within the cornea, respiratory tract, and/or gastrointestinal tract and vary in size from a few millimeters to large bullae. Dystrophic epidermolysis bullosa (DEB) is 1 of 4 main types of epidermolysis bullosa that also include Epidermolysis Bullosa Simplex (EBS) with blistering in the epidermis, Junctional Epidermolysis Bullosa (EBJ) with blistering within the basement membrane zone, and Kindler syndrome with blistering at multiple levels and variable photosensitivity.1,2
DEB is associated with blisters in the upper dermis at the sublamina densa level that eventually heal with scarring and may result in milia formation. Patients may also have extracutaneous findings. There are 2 forms of DEB, subdivided according to autosomal dominant and recessive inheritance. The dominant form has recently been categorized as localized (previously nails only, pretibial, and acral dominant) and intermediate (previously generalized dominant) types, while recessive DEB has been divided into intermediate and severe classifications.1
DDEB presents at birth or early in life and is generally less severe. The estimated prevalence of DDEB in the United States is 1.49 per 1 million individuals and incidence is 2.12 per 1 million live births.3 Blisters are localized on the elbows, knees, and dorsum of the hands and feet. Nail dystrophy and loss are frequent. Patients with recessive DEB often have more widespread blistering and serious systemic findings. Oral lesions can later lead to ankyloglossia and microstomia, and patients may suffer from dental caries and loss of teeth.4,5 Gastrointestinal complications include esophageal strictures, dysphagia, gastroesophageal reflux, and intestinal erosions that can contribute to development of anemia and impaired growth.6,7 As time progresses, patients have high prevalence of osteopenia and osteoporosis.8

Differential diagnosis
The differential diagnosis for disorders of blistering and skin fragility can be divided into inherited and acquired bullous disorders. Inherited bullous disorders are characterized as epidermolysis bullosa and its subtypes, epidermolytic ichthyosis, incontinentia pigmenti, and ankyloblepharon-ectodermal dysplasia-clefting syndrome. Acquired bullous disorders consist of nonautoimmune and autoimmune etiologies. Nonautoimmune conditions include thermal burns, bullous mastocytosis, and infections such as bullous impetigo, intrauterine herpes simplex virus, and congenital varicella. Stevens-Johnson syndrome and toxic epidermal necrolysis, both potentially life-threatening, are other possibilities that can be determined from the patient’s history and inquiring about potential inciting exposures such as recent medication use. Autoimmune conditions, though rare in infants, include linear IgA bullous dermatosis, pemphigus vulgaris, pemphigus foliaceus, epidermolysis bullosa acquisita, and bullous systemic lupus erythematosus. Maternal history of autoimmune blistering disorders is an important predisposing factor. Histopathological and immunofluorescence studies are useful for detecting autoantibodies that underlie autoimmune bullous disorders.
Definitive diagnosis includes biopsy with immunofluorescence antigen mapping. Further genetic testing can be pursued. Mutations in the COL7A1 gene, which encodes collagen VII and is comprised of 3 polypeptide chains that form the anchoring filaments, have been implicated in DEB.2,9,10 Our patient’s diagnosis of DEB was confirmed with these 2 tests.
Treatment
The treatment regimen for DEB includes supportive care individualized to the patient’s disease severity. For children, the goal is to maintain health and growth, alleviate symptoms and pain, and provide support for caregivers. Topical emollients and nonadherent dressings help protect fragile skin. Avoidance of minor trauma and rubbing is necessary, and parents can place padding to shield areas of the body more prone to exposure. Patients have increased susceptibility to superimposed bacterial infections due to their damaged skin barrier and may require systemic antibiotics for infections.
Given the potential multiorgan involvement of the disease and substantial impact on quality of life, optimizing medical care for patients suffering from DEB requires a multidisciplinary approach. In the long term, patients may require further surgical or medical interventions to address cutaneous scarring and extradermatological symptoms. For instance, patients with esophageal strictures can be addressed with balloon dilation, which provides lasting improvement in dysphagia.11 Those whose injuries have led to severe limb deformities, such as hand contractures and pseudosyndactyly, have functional benefit from surgical correction.12,13 Dental implants provide useful oral rehabilitation for patients who have required teeth extraction or who have had periodontal disease.4 Psychosocial support can be very valuable, as patients with DEB often report tremendous emotional burdens and difficulty coping with the rarity and lifelong nature of the disease.14-16
Clinical course
Our patient received supportive therapy, and his wounds healed well under appropriate wound care. Of note, his mother was experienced in managing these wounds from her own experience and felt well prepared in caring for her infant. Continued symptomatic treatment helped prevent potential complications and further injury. Over time, the patient’s feeding, growth, and development progressed well. Most children with epidermolysis bullosa who receive timely, comprehensive medical therapy and who survive beyond the first 1 to 2 years will also survive into adulthood.17 Due to the risk of possible comorbidities, however, children benefit from continued care; for instance, patients with recessive DEB have an increased risk of squamous cell carcinoma (SCC) and will require future skin surveillance.18 The pathophysiology of SCC in patients with DEB is complex and remains an area of active investigation; it is likely multifactorial, resulting from increased susceptibility in the skin barrier, inflammation, and impaired healing responses. Ultimately, due to the complexity of this disease, long-term follow-up with dermatologists and other medical specialists is essential.
References:
1. Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183(4):614-627. doi:10.1111/bjd.18921
2. Fine JD, Bruckner-Tuderman L, Eady RAJ, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70(6):1103-1126. doi:10.1016/j.jaad.2014.01.903
3. Fine JD. Epidemiology of inherited epidermolysis bullosa based on incidence and prevalence estimates from the National Epidermolysis Bullosa Registry. JAMA Dermatol. 2016;152(11):1231-1238. doi:10.1001/jamadermatol.2016.2473
4. Peñarrocha-Oltra D, Agustín-Panadero R, Serra-Pastor B, Peñarrocha-Diago M, Peñarrocha-Diago M. Oral rehabilitation with dental implants in patients with recessive dystrophic epidermolysis bullosa: a retrospective study with 2-15 years of follow-up. Med Oral Patol Oral Cir Bucal. 2020;25(2):e262-e267. doi:10.4317/medoral.23331
5. Serrano-Martínez MC, Bagán JV, Silvestre FJ, Viguer MT. Oral lesions in recessive dystrophic epidermolysis bullosa. Oral Dis. 2003;9(5):264-268. doi:10.1034/j.1601-0825.2003.03971.x
6. Fine JD, Johnson LB, Weiner M, Suchindran C. Gastrointestinal complications of inherited epidermolysis bullosa: cumulative experience of the National Epidermolysis Bullosa Registry. J Pediatr Gastroenterol Nutr. 2008;46(2):147-158. doi:10.1097/MPG.0b013e31812f5667
7. Freeman EB, Köglmeier J, Martinez AE. Gastrointestinal complications of epidermolysis bullosa in children. Br J Dermatol. 2008;158(6):1308-1314. doi:10.1111/j.1365-2133.2008.08507.x
8. Kang M, Chen JSC, Radjenovic M, Yang A, Feng GHY, Murrell DF. An analysis of the prevalence of osteoporosis and osteopenia in patients with epidermolysis bullosa: a cross-sectional study. Exp Dermatol. 2021;30(11):1675-1685. doi:10.111/exd.14252
9. McGrath JA, Ishida-Yamamoto A, O'Grady A, Leigh IM, Eady RA. Structural variations in anchoring fibrils in dystrophic epidermolysis bullosa: correlation with type VII collagen expression. J Invest Dermatol. 1993;100(4):366-372. doi:10.1111/1523-1747.ep12471830
10. Bruckner-Tuderman L, Mitsuhashi Y, Schnyder UW, Brukner P. Anchoring fibrils and type VII collagen are absent from skin in severe recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 1989;93(1):3-9. doi:10.1111/1523-1747.ep12277331
11. Spiliopoulos S, Sabharwal T, Krokidis M, et al. Fluoroscopically guided dilation of esophageal strictures in patients with dystrophic epidermolysis bullosa: long-term results. AJR Am J Roentgenol. 2012;199(1):208-212. doi:10.2214/AJR.11.8159
12. Zhou X, Zhang Y, Zhao M, et al. Surgical management of hand deformities in patients with recessive dystrophic epidermolysis bullosa. J Plast Surg Hand Surg. 2020;54(1):33-39. doi:10.1080/2000656X.2019.1661846
13. Lembo F, Parisi D, Cecchino LR, Ciancio F, Innocenti A, Portincasa A. Release of pseudosyndactyly in recessive dystrophic epidermolysis bullosa using a dermal regeneration template glove: the Foggia experience. Orphanet J Rare Dis. 2021;16(1):52. doi:10.1186/s13023-021-01697-5
14. Dures E, Morris M, Gleeson K, Rumsey N. The psychosocial impact of epidermolysis bullosa. Qual Health Res. 2011;21(6):771-782. doi:10.1177/1049732311400431
15. Bruckner AL, Losow M, Wisk J, et al. The challenges of living with and managing epidermolysis bullosa: insights from patients and caregivers. Orphanet J Rare Dis. 2020;15(1):1. doi:10.1186/s13023-019-1279-y
16. Mauritz P, Jonkman MF, Visser SS, Finkenauer C, Duipmans JC, Hagedoorn M. Impact of painful wound care in epidermolysis bullosa during childhood: an interview study with adult patients and parents. Acta Derm Venereol. 2019;99(9):783-788. doi:10.2340/00015555-3179
17. Fine JD, Johnson LB, Weiner M, Suchindran C. Cause-specific risks of childhood death in inherited epidermolysis bullosa. J Pediatr. 2008;152(2):276-280. doi:10.1016/j.jpeds.2007.06.039
18. Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. J Am Acad Dermatol. 2009;60(2):203-211. doi:10.1016/j.jaad.2008.09.035
For more from the July, 2023 issue of Contemporary Pediatrics®, click here.











Newsletter
Access practical, evidence-based guidance to support better care for our youngest patients. Join our email list for the latest clinical updates.









Recognize & Refer: Hemangiomas in pediatrics
July 17th 2019Contemporary Pediatrics sits down exclusively with Sheila Fallon Friedlander, MD, a professor dermatology and pediatrics, to discuss the one key condition for which she believes community pediatricians should be especially aware-hemangiomas.




