
|Articles|December 1, 2008
Stevens-Johnson Syndrome
A 13-year-old boy was brought to the emergency department (ED) with a generalized itchy rash of 2 days' duration. For the past 3 days, he had dry, itchy eyes with a purulent discharge (Figure 1) and nonbilious emesis 2 or 3 times per day, with some blood streaks in the vomitus on the third day of illness.
Advertisement
A 13-year-old boy was brought to the emergency department (ED) with a generalized itchy rash of 2 days' duration. For the past 3 days, he had dry, itchy eyes with a purulent discharge (Figure 1) and nonbilious emesis 2 or 3 times per day, with some blood streaks in the vomitus on the third day of illness. Chills developed on the third day, but the patient's temperature was not measured. His lips were swollen and cracked (Figure 2), which made it painful to eat or drink. About 6 months earlier, he had a similar episode that resolved with application of a topical cream to the lips. He also had painful urination without urgency or penile discharge. He denied recent use of medications and had no ill family members. He had been well before the onset of symptoms.
Figure 1 – About 30% of children with Stevens-Johnson syndrome have ocular involvement. This 13-year-old boy had mucopurulent conjunctivitis with episcleritis as a manifestation of the disease.
Figure 2 – Mucositis of the lips in this teenager was a manifestation of Stevens-Johnson syndrome. Oral lesions are the most common type of mucosal involvement in affected patients. They are extremely painful and limit oral intake.
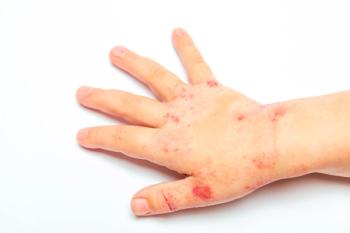
The teenager was apathetic, mildly dehydrated, and appeared to be in pain. Oral temperature was 39.6ºC (103.2ºF); respiration rate, 20 breaths per minute; heart rate, 108 beats per minute; and blood pressure, 108/63 mm Hg. Physical examination revealed injection of the conjunctivae with sticky yellow discharge, swollen lips with erosion of the mucosa, hemorrhagic crusting, intraoral ulcers, dry oral mucous membranes, ulceration of the head of the penis, and a generalized rash that consisted of target lesions with central erosions.
The white blood cell count was normal (6700/μL), without a left shift; hemoglobin level was 14.3 g/dL. Results of coagulation studies were normal. The erythrocyte sedimentation rate and C-reactive protein level were markedly elevated (107 mm/h and 33.78 mg/L, respectively). Serum electrolyte levels were normal, blood urea nitrogen level was elevated (33 mg/dL), and creatinine level was 1.3 mg/dL. Liver function test results were normal; the albumin level was slightly below normal (3.3 g/dL). Urinalysis results showed a specific gravity of 1.028 and 1+ protein; all other parameters were within normal range. IgA was 183 mg/dL. Direct fluorescent antibody studies of lip vesicles for herpes simplex virus types 1 and 2 were negative. Fluorescein examination of the eye demonstrated no corneal uptake of dye. A blood sample for culture was obtained.
In the ED, the patient received intravenous saline, ceftriaxone (a single dose), acyclovir, diphenhydramine, and analgesia. A topical analgesic was applied to the ulcerated mouth, and emollients were applied to the rash. The ophthalmology consultant diagnosed mucopurulent conjunctivitis with episcleritis and recommended lubrication with artificial tears and erythromycin ophthalmic ointment. The dermatology consultant performed a biopsy of a target lesion, which demonstrated interface dermatitis with vacuolation of the basal layer and rare dyskeratotic cells in the epidermis suggestive of Stevens- Johnson syndrome (SJS) or erythema multiforme (EM).
Serological test results were negative for Mycoplasma; herpes simplex virus; Epstein-Barr virus; Cytomegalovirus; HIV; hepatitis A, B, and C viruses; and Treponema pallidum. Blood culture was negative.
Because of persistent vomiting and a steadily increasing elevated lipase level (from 713 U/L to a maximum of 1589 U/L), abdominal ultrasonography was performed. The size of the pancreas was at the upper limits of normal; there was a small amount of free fluid in the abdomen.
During the patient's 10-day hospitalization, intravenous hydration was maintained until he was able to tolerate oral feeds. At discharge, the eye and skin lesions had completely resolved and the oral ulcers had almost completely healed. The final diagnosis was SJS and acute pancreatitis.(Discussion on next page.)
EM MAJOR: AN OVERVIEW
SJS and toxic epidermal necrolysis (TEN) are severe mucocutaneous blistering disorders of acute onset that are associated with considerable morbidity and life-threatening complications. Like EM minor, both SJS and TEN may have oral mucosal lesions and ulcerated lesions; however, SJS and TEN are distinguished from EM by the involvement of 2 or more mucosal surfaces, more widespread lesions, systemic illness, and more extensive epidermal involvement on biopsy.
von Hebra1 first described EM as a self-limited disease in healthy young adults that is most commonly caused by herpes simplex virus, has the classic iris or target lesion, and is rarely associated with fever or systemic symptoms. Stevens and Johnson2 later described a severe, generalized skin infection, prolonged high fever, purulent conjunctivitis, and severe oral stomatitis in 2 children. They excluded the diagnosis of EM in these 2 children because of the presence of atypical target lesions and prolonged systemic symptoms.
The term "EM minor" became synonymous with von Hebra disease and "EM major" (EM with involvement of 2 mucous membranes) with SJS. TEN, first described by Lyell,3 is currently considered to be a severe form of SJS caused by a similar spectrum of drugs and characterized by full-thickness epidermal loss over a large extent of the body surface. The current thinking is that there are 2 groups of diseases:
•EM and bullous EM are primarily caused by herpes simplex virus, present with classic target lesions in an acral distribution, and have a benign course.
•SJS and TEN are often druginduced, are characterized by the presence of atypical target lesions and purpuric macules, and cause severe morbidity.
The current classification of acute severe bullous disorders comes from Bastuji-Garin and colleagues4 and is based on the extent of body surface involvement and type of skin lesions. SJS has an estimated incidence of2.6 cases per million per year5 and affects all age-groups.
ETIOLOGY
Drugs are the most common cause of SJS and TEN,6 although infectious agents and immunizations have also been implicated. Drugs that frequently cause SJS include antibiotics (particularly sulfonamides, aminopenicillins, and cephalosporins) and aromatic anticonvulsants- phenobarbital, phenytoin, and carbamazepine. 7 While NSAIDs and antigout medications frequently cause SJS in adults, they are rarely implicated in children. Drug-induced SJS typically occurs between 1 and 3 weeks of initiation of drug therapy. Persons with systemic lupus erythematosus and those who are infected with HIV are more susceptible to drug-induced SJS.
Among infectious causes, herpes simplex virus and Mycoplasma pneumoniae are most strongly associated with SJS. Epstein-Barr virus, Cytomegalovirus, and streptococcal infections are also associated with SJS. Herpes simplex virus 1 infection was the suspected cause of SJS in this patient based on the presence and previous episode of lip lesions. However, given the negative serological results, this could not be proved conclusively.
PATHOGENESIS
SJS is believed to be an immunological disorder that causes perivasculitis of the superficial dermal vessels. Immunofluorescent examination of early skin lesions in SJS demonstrate IgM and C3 deposits in the vascular walls in most patients. Other histological changes include full-thickness necrosis of the epidermis and a scant mononuclear cell in the dermis. The dermoepidermal junction shows changes that range from vacuolar alteration to subepidermal blister formation. In the drug-induced form, it is believed that full-thickness epidermal necrosis is triggered by accumulation of the drug metabolites in the epidermis. Patients and their first-degree relatives are believed to have genetic defects in the drug metabolic pathway that leads to accumulation of the drug metabolites. Persons with HLAB12 (HLA-Bw44) antigen are at greater risk for the development of SJS.8 CD8+ T-cell–mediated cytotoxicity is believed to be responsible for the epidermal necrosis in TEN.
CLINICAL FEATURES
SJS is usually preceded by a prodrome of fever, sore throat, and generalized malaise that lasts for several days and is often misdiagnosed as an infectious illness for which an antibiotic is prescribed. In a large case series from Toronto, 61% of children had mucous membrane involvement, most commonly oral, genital, and anal.9 Of those children, 95% had oral lesions that ranged from isolated vesicles or bullae to swelling, blistering, and ulceration of both lips. The mouth lesions seen in SJS are extremely painful and limit oral intake, as was the case in this patient.
Genital lesions include vesicobullous vulvovaginitis, ulcers, and anal and urethral erosions. About 30% of children have ocular involvement in the form of hemorrhagic conjunctivitis, corneal ulceration, blepharitis, or scleritis.9 These can lead to corneal erosions, pseudomembrane formation, and adhesions. Rarely, other mucous membranes, such as the esophagus, intestinal tract, respiratory epithelium, and nasal cavity, can also be affected.
The rash of SJS usually begins as purpuric macules that evolve within days into atypical targetlike bullae and erosions of the face, trunk, limbs, palms, and soles. The scalp is usually spared. Typical target lesions are usually less than 3 cm in diameter and round with a well-defined border. They have 3 zones with 2 concentric rings, 1 of which is palpably edematous and paler than the central disk. Atypical target lesions are also round, edematous, palpable lesions but have only 2 zones and an ill-defined border.
The rash tends to be more severe and extensive in patients with drug-induced SJS. It can progress to sheetlike loss of epidermis, especially in cases of TEN. The Nikolsky sign (formation of a blister with peeling of the superficial layers of skin from the lower layers, leaving wet, red, painful areas on application of horizontal, tangential pressure to the skin) is usually positive in TEN. The average duration of illness is usually 2 weeks. Resolution and healing occur between 1 and 2 weeks.
(Discussion continues on next page.)
LABORATORY FINDINGS
SJS is usually a clinical diagnosis. Abnormal laboratory findings may include an elevated sedimentation rate, hypoalbuminemia (as in this patient), mildly elevated liver enzyme levels, microscopic hematuria, and mild leukocytosis. Leukopenia is considered a poor prognostic sign and may indicate superinfection and sepsis. Serological test results may be positive in Mycoplasma-induced SJS.
A skin biopsy is indicated only when the diagnosis is uncertain. Histopathological findings include a mononuclear perivascular cell infiltrate in the dermis, basal layer edema, subepidermal blister formation, and epidermal cell necrosis. TEN is characterized by epidermal necrosis with minimal evidence of inflammation.
COMPLICATIONS AND SEQUELAE
A mortality rate of up to 5% has been reported with SJS.10 Patient age and extent of skin detachment have been proposed as the main prognostic factors. Early withdrawal of the causative agent, when one is involved, may decrease mortality. 11 The most common complication is sepsis (most often caused by Staphylococcus aureus followed by Pseudomonas aeruginosa).
Hypovolemic shock can occur from fluid loss and electrolyte imbalance secondary to extensive skin detachment. Sloughing and edema of the respiratory epithelium may lead to upper airway obstruction, pneumonia, or diffuse interstitial pulmonary disease. Urethral erosions can cause acute urinary retention, especially in younger children. GI erosions can lead to upper GI bleeding and bloody diarrhea. Less common sequelae include cutaneous scarring, palpebral synechiae with permanent visual impairment, and bronchiolitis obliterans.
In a review of the literature, there was no reported association between pancreatitis and SJS, although some of the drugs that cause SJS (such as valproic acid) also cause pancreatitis. We do not believe that the pancreatitis led to SJS in this patient.
MANAGEMENT
SJS and TEN in children are dermatological emergencies. Diagnosis should prompt immediate discontinuation of all potential causative drugs. Older children with proven Mycoplasma infection can be treated with either an oral macrolide or oral doxycycline. Patients with extensive blistering or skin loss require care at a pediatric burn center. Those with extensive skin detachment require reverse barrier isolation and a sterile environment. Consultation with an ophthalmologist and dermatologist is recommended.
Supportive measures include isolation, fluid and electrolyte balance, nutritional support, eye and mouth care, pain management, and nonadherent protective dressings for skin lesions.12 Because of poor oral intake, many children require nutritional support with either a nasogastric tube or parenteral alimentation for 1 to 2 weeks. While surgeons advocate extensive debridement, as in a burn patient, dermatologists recommend debridement of necrotic and peeled skin only. The value of topical antibiotics is unproved. No particular type of dressing (synthetic, biological, or petrolatum gauze) has been proved superior. Children often require an opioid analgesic for dressing changes.
Other than good supportive care, there is no effective and universally accepted treatment of SJS. Although prevention and early treatment of sepsis is indicated, prophylactic antibiotics are not recommended.
The role of corticosteroids in the treatment of SJS or TEN is controversial. Although earlier retrospective studies reported a longer hospital stay and a higher incidence of complications in children with SJS who were treated with corticosteroids, 13 other studies reported an outstanding success rate with early and prompt treatment with highdose corticosteroids.14 To date, no randomized controlled studies have shown a benefit from their use in the treatment of SJS. Moreover, SJS can be managed successfully with supportive care alone.12
Although some case series reported improved outcomes in patients with SJS after a single infusion of intravenous immunoglobulin (IVIG),15,16 a prospective study failed to show reduction in mortality or progression of disease.17 Randomized controlled trials with large numbers of patients are needed before IVIG can be recommended as a standard treatment for children with SJS.
References:
REFERENCES:1. von Hebra F. On Diseases of the Skin Including the Exanthemata. Fagge CH, trans. London: New Sydenham Society; 1866:285-289.
2. Stevens AM, Johnson FC. A new eruptive fever associated with stomatitis and ophthalmia: report of two cases in children. Am J Dis Child. 1922;24: 526-533.
3. Lyell A. Toxic epidermal necrolysis. An eruption resembling scalding of the skin. Br J Dermatol. 1956;68:355-361.
4. Bastuji-Garin S, Rzany B, Stern RS, et al. Clinical classification of cases of toxic epidermal necrolysis, Stevens-Johnson syndrome, and erythema multiforme. Arch Dermatol. 1993;129:92-96.
5. Schöpf E, Stühmer A, Rzany A, et al. Toxic epidermal necrolysis and Stevens-Johnson syndrome. An epidemiologic study from West Germany. Arch Dermatol. 1991;127:839-842.
6. Roujeau JC, Kelly JP, Naldi L, et al. Medication use and the risk of Stevens-Johnson syndrome or toxic epidermal necrolysis. N Engl J Med. 1995;333: 1600-1607.
7. Prendiville J. Stevens-Johnson syndrome and toxic epidermal necrolysis. Adv Dermatol. 2002;18: 157-173.
8. Mondino BJ, Brown SI, Biglan AW, et al. HLA antigens in Stevens-Johnson syndrome with ocular involvement. Arch Ophthalmol. 1982;100:1453-1454.
9. Forman R, Koren G, Shear NH. Erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis in children: a review of 10 years' experience. Drug Saf. 2002;25:965-972.
10. Revuz J, Penso D, Roujeau JC, et al. Toxic epidermal necrolysis: clinical findings and prognosis in 87 patients.Arch Dermatol. 1987;123:1160-1165.
11. Garcia-Doval I, LeCleach L, Bocquet H, et al. Toxic epidermal necrolysis and Stevens-Johnson syndrome: does early withdrawal of causative drugs decrease the risk of death? Arch Dermatol. 2000; 136:323-327.
12. Prendiville JS, Hebert AA, Greenwald MJ, Esterly NB. Management of Stevens-Johnson syndrome and toxic epidermal necrolysis in children. J Pediatr. 1989;115:881-887.
13. Kim PS, Goldfarb IW, Gaisford JC, et al. Stevens- Johnson syndrome and toxic epidermal necrolysis: a pathophysiologic review with recommendations for a treatment protocol. J Burn Care Rehabil. 1983; 4:91-100.
14. Patterson R, Miller M, Kaplan M, et al. Effectiveness of early therapy with corticosteroids in Stevens- Johnson syndrome: experience with 41 cases and a hypothesis regarding pathogenesis. Ann Allergy. 1994;73:27-34.
15. Metry DW, Jung P, Levy ML. Use of intravenous immunoglobulin in children with Stevens-Johnson syndrome and toxic epidermal necrolysis. Pediatrics. 2003;112:1430-1436.
16. Prins C, Vittorio C, Padilla RS, et al. Effect of high-dose intravenous immunoglobulin therapy in Stevens-Johnson syndrome: a retrospective, multicenter study. Dermatology. 2003;207:96-99.
17. Bachot N, Revuz J, Roujeau JC. Intravenous immunoglobulin treatment for Stevens-Johnson syndrome and toxic epidermal necrolysis: a prospective noncomparative study showing no benefit on mortality or progression. Arch Dermatol. 2003;139:33-36.
Advertisement
Related Content
Advertisement



Advertisement
Advertisement
Trending on Contemporary Pediatrics
1
FDA approves acetaminophen-naproxen sodium combination tablet for ages 12 and older
2
US measles cases surpass 2025 total, hit highest level since 1991
3
Pooled phase 3 data show early, consistent tapinarof cream responses across pediatric age groups in AD
4
Individualized gene therapy shows early promise for SCN2A-related epileptic encephalopathy
5