
|Articles|October 1, 2006
- Consultant for Pediatricians Vol 5 No 10
- Volume 5
- Issue 10
Photoclinic: Incontinentia Pigmenti
Photoclinic: Incontinentia Pigmenti
Advertisement
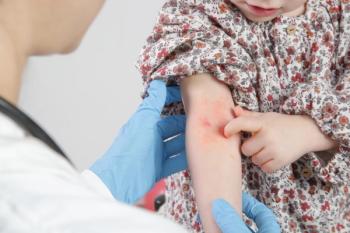
The skin eruption on this 2-month-old infant had been present since birth. The mother was told it was a "newborn rash." At a well-child visit a week after the baby was born, the primary care physician referred the infant to a dermatologist, who diagnosed "an infection" and prescribed cephalexin and a topical antibiotic.
When these medications did not alter the rash, the mother sought the opinion of a pediatrician. At that visit, she reported that the dermatitis had spread initially, but that some lesions had healed and faded. The rash seemed to have no associated pruritus or pain and had persisted despite antibiotic therapy.
The infant had been born at 38 weeks gestation via vaginal delivery without complications to a gravida 2, para 1, aborta 1 mother. She had a spontaneous abortion at about 2 to 3 months' gestation of her first pregnancy. During the current pregnancy, the mother worked in an animal shelter where she had contact with cats. She had no history of sexually transmitted diseases.
The infant was otherwise healthy. The eyes showed normal red reflex and no cataracts. Cardiac examination findings were normal; there was no appreciable murmur. The rash consisted of multiple large, vesicular patches with coalesced, erythematous bases--some in linear groups--mainly on the upper and lower extremities. Verrucous, hyperpigmented patches were noted on the thighs, and reddish, hyperpigmented linear arrays were noted on the flanks and axillae bilaterally. A whorl of lesions extended up the right flank to the right abdominal wall.
Paul E. Trombley, MD, of Presbyterian Española Hospital in New Mexico made the presumptive diagnosis of incontinentia pigmenti. Results of a complete blood cell count showed leukocytosis, with 46% eosinophils. A workup for TORCH syndrome (infection with toxoplasmosis, other infections, rubella, cytomegalovirus infection, and herpes simplex) was negative. A NEMO gene assay was positive for the Xq28 deletion, which confirmed the diagnosis.
On further questioning, the mother revealed that she did not know the sex of the first fetus, but it was suspected to have been a male child.
Incontinentia pigmenti (Bloch-Sulzberger syndrome) affects the skin, CNS, eyes, and skeletal systems. This rare disorder manifests shortly after birth (typically within the first 2 weeks of life) and predominantly affects females.1 It is thought to be fatal in prenatal, hemizygous males. The extremely rare case in a male newborn is believed to be the result of spontaneous mutations.2
Two gene loci for incontinentia pigmenti have been identified: IP1 at Xp11.21 and IP2 at Xq28. The mutation is suspected to produce a failure of immune tolerance in ectodermal tissues that causes an autoimmune-like reaction in heterozygous girls and a fatal graft versus host-like disease in boys.2
The 4 distinct cutaneous stages of incontinentia pigmenti frequently overlap. The first stage includes inflammatory vesicles or bullae on the trunk and extremities. Dermal biopsy during this stage shows inflammatory infiltrates of polymorphonuclear leukocytes, lymphocytes, and eosinophils; prominent peripheral eosinophilic leukocytosis occurs in 74% of patients, as was noted in the case. The second stage is characterized by irregular, linear, warty or verrucous lesions on one or more extremities. During this stage, biopsy may demonstrate acanthosis, irregular papillomatosis, hyperkeratosis, basal cell vacuolation with decreased melanin content, and a mild chronic inflammatory infiltrate. The third stage is characterized by bands of brown to bluish gray whirling or linear patterns on the trunk and extremities. During this stage, biopsy may demonstrate extensive melanin deposition. In the last stage, skin changes are characterized by subtle, streaky, hypopigmented scars that may be the only residual cutaneous findings in adult women. Biopsy during this stage demonstrates decreased melanin and skin appendages within the atrophic scars.2,3
The differential diagnosis of incontinentia pigmenti changes with each stage:
Inflammatory stage: epidermolysis bullosa, linear IgA dermatosis, and childhood bullous pemphigus
Verrucous stage: linear epidermal nevus
Hyperpigmented stage: lichen planus or lichen nitidus
Hypopigmented late stage: the lesions may appear as hypomelanosis of Ito2
About 30% of patients with incontinentia pigmenti have CNS manifestations, such as seizures (3.3%), mental retardation (16%), and spastic abnormalities (13%). Up to 35% of patients may have ophthalmologic complications, including strabismus, cataracts, optic atrophy, and microphthalmia. Alopecia may occur in up to 38% of patients, and another 7% may have nail dystrophy. Abnormal dentition--the most common finding--manifests as delayed dentition or pegged or conical teeth in 64% of patients.2,3 Cardiac and skeletal abnormalities, including hemivertebrae, kyphoscoliosis, an extra rib, syndactyly, hemiatrophy, and short arms and legs, rarely have been reported.1
Up to 80% of all patients with incontinentia pigmenti have systemic disease. Those who have seizures within the first week of life have a poor prognosis. Normal developmental milestones and the absence of seizures appear to predict a good prognosis. No treatment is necessary for this disorder; however, genetic counseling is advisable.2
References:
REFERENCES:
1.
Jones K, ed.
Smith's Recognizable Patterns of Human Malformations.
5th ed. Philadelphia: WB Saunders; 1997:502-503.
2.
Kane K, Bissonette J, eds.
Color Atlas & Synopsis of Pediatric Dermatology.
New York: McGraw-Hill; 2002:292-294.
3.
Zitelli BJ, Davis H, eds.
Atlas of Pediatric Physical Diagnosis.
3rd ed. St Louis: Mosby-Wolfe; 1997: 205-215.
Articles in this issue
almost 20 years ago
Photoclinic: Hereditary Hemorrhagic Telangiectasiaalmost 20 years ago
e-Photo Quiz: Answer to Last Month’s Online Challenge - Orbital cellulitisalmost 20 years ago
Diabetic Ketoacidosis in Children:almost 20 years ago
Adolecent Medicine Update:almost 20 years ago
A Collage of Hereditary Childhood Disordersalmost 20 years ago
Orbital Abscessalmost 20 years ago
Herpes Zoster (Shingles) in a Teenageralmost 20 years ago
Case in Point: Night Terrorsalmost 20 years ago
Editors Commentary: Three in One . . .almost 20 years ago
Anaphylactic Reaction to Milk in an InfantAdvertisement
Related Content
Advertisement

Advertisement
Advertisement
Trending on Contemporary Pediatrics
1
FAQ: What does the AAP's updated drowning prevention policy mean for pediatric practice?
2
Study finds no association between neonatal circumcision and autism diagnosis
3
FDA news in pediatrics: July 2026
4
FDA accepts application to expand clesrovimab-cfor use to high-risk children in second RSV season
5